




2. 中国科学院 研究生院, 北京100049
2. Graduate University of Chinese Academy of Sciences, Beijing 100049, P.R.China
Calcium phosphates have been widely studied inside the scope of biomaterials [1,2,3] and clinically used in orthopedic [3,4,5] and bone repair [6,7,8] surgeries,mainly because of their chemical similarities with the inorganic part of bone and teeth. Apatite (HAP) and Pure β-tricalcium phosphate (β-TCP)[2,9] are two of the most widespread used calcium phosphate-based biomaterials. One is due to its biocompatibility[10] and the other due to its advanced bioresorpbility[11, 12]. Although HAP is the most thermodynamically favored calcium phosphate and have been considered to be identical with the inorganic composition of bones or tooth,its low solubility makes it hard to be degraded in the bone defect repair[13]. On the contrary,β-TCP has higher solubility[14, 15] because of its high surface energy and can be modulated to match the rate of new bone formation.
In general,β-TCP is synthesized by adopting the aqueous precipitation method[16,17,18,19]. However,the precipitate instantaneously derived from solution is always calcium deficient apatite (Ca-dHAP) Ca10-x(PO4)6-x(HPO4)x(OH)2-x,with 0≤x≤1. The x value varies according to the substitution of phosphate ions (PO43-) by hydrogenophosphate ones (HPO42-) or carbonate (CO32-),which leads to a continuous variation of the Ca/P atomic ratio between 1.5 and 1.667 of Ca-dHAP and the final calcined calcium phosphates[20]. Raynaud[21, 22] et al. have systematically studied the final Ca/P ratio of the calcined precipitate by strictly controlling the pH,temperature,and the initial Ca/P of the precursor. They paid more attention to the final precipitates with Ca/P between 1.5 and 1.667 under the constant pH and temperature reaction conditions. Meanwhile they also noticed that the synthesis of pure apatitic TCP (Ca/P=1.50) appeared less reproducible and the biphasic powders with Ca/P<1.5 were easily obtained as the initial Ca/P was below 1.5.
In bone,calcium and phosphate ions are induced by the non-collagenous proteins to precipitate and deposit on the collagen template. Gelatin is derived from collagen by thermal,physical or chemical degradation of collagen,and can provide a high surface density of anionic functional groups. Some in vitro studies indicated that the anionic groups,such as the phosphate and carboxyl groups,on the surface of organic molecules could attract calcium ions by electrostatic force as nucleating agents for apatite crystals[23, 24]. In this paper,we adopt this biomineralization strategy and are concerned with the initial Ca/P ratio of 1.5. Furthermore,the effect of gelatin on the formation of Ca-dHAP and its subsequent phase transformation were studied. 1 Materials and Methods
The conventional wet chemical precipitation method was employed to synthesize β-TCP,using calcium nitrate tetrahydrate (Ca (NO3)2·4H2O) (Institute of Jinke Industry of Fine Chemicals,Tianjin,China) and diammonium hydrogenphosphate ((NH4)2HPO4) (Shantou Xilong Chemical Factory,Guangdong,China) as the starting precursors. Ammonium hydroxide (NH3·H2O) (Beijing Chemical Works,China) was used to control and regulate the pH of the reaction. All reagents used were of chemical analysis grade. 1.1 Synthesis of gelatin-induced calcium phosphates
0.04 mol (NH4)2HPO4 and 0.06 mol Ca (NO3)2 ·4H2O were first dissolved in 40 mL and 30mL de-ionized water,respectively. After dissolved,ammonium hydroxide was added into these two solutions to adjust the pH both above 10.8,monitored with a pH stat (PHS-3BW pH/mV meter,LIDA Instrument Factory,Shanghai,China). Then a certain volume of gelatin (de-ionized gelatin,296 bloom,Baotou Dongbao Biotechnology Co. Ltd.,China) solution was added into the (NH4)2HPO4 solution (pH did not change greatly). Next,Ca (NO3)2 ·4H2O solution was added dropwise to the pre-mixtured (NH4)2HPO4-gelatin mixtures over 60 min to produce a white slurry. Throughout the mixing process,the system was vigorously stirred at room temperature and the pH was maintained above 10.8 with adding ammonium hydroxide. The slurry was then stirred for 12 h,followed by washing with de-ionized water,and then washed with pure ethanol to improve the dispersion characteristics. After washing,the remaining liquid was removed by vacuum filtration and the obtained precipitate was dried at 80 ℃ for 24 h. The dried precipitates were calcined at 800 ℃ for 2 h to produce the β-TCP product. All prepared powders were preserved in sealed plastic bags for characterization.
Sample notations were coded according to the amount of gelatin added into the solution,such as: (a) for 0.00 g gelatin,(b) for 0.020 g gelatin,(c) for 0.040 g gelatin,(d) for 0.20 g gelatin,(e) for 0.50 g gelatin.
X-ray diffraction analysis was carried out by means of a D8 Focus (Bruker,Germany) powder diffractometer and CuKα radiation was used (40 mA,40 kV). The 2θ range was from 10° to 50° at a scanning speed of 0.01°/s with an increment of 0.02 step/s. The crystallite sizes are calculated from XRD data using the Debye-Scherrer equation D=Kλ/βcosθ,Where D is the crystallite size,as calculated for the (hkl) reflection; K is the shape factor (a value of 0.89 was used); λ is the wavelength of used X-rays (0.15406 nm); and θ is the Bragg’s diffraction angle. The crystallite sizes were obtained by measuring the width at half the maximum intensity β of the peak (002) plane,representative of the crystallites along the c axis; and of the peak (300) plane,representative of the crystallites along the α axis.
The FT-IR absorption analysis was carried out on an Excalibur 3100 spectrometer (Varian,USA) in the range of 400 cm-14000 cm-1,using KBr as pellets.
The specific surface area of the as-prepared powders was measured by the BET method on Quadrasorb SI-MP (Quantachrome,USA).
Morphological investigation and semi-quantitative determination of the element analysis of the prepared and calcined powders were observed on a transmission electron microscope (JEOL JEM-2100F TEM,Japan) at an acceleration voltage of 200 kV.
The dried powders were also subjected to thermal analysis with a heating rate of 10 ℃/min between room temperature and 800 ℃ under the static air atmosphere (Diamond TG/DTA,PerkinElmer,USA) to analyze the thermal behavior during heating. 2 Results and Discussions
According to Fig.1,pure β-TCP is obtained at 800 ℃ when the gelatin concentration exceeds 0.22%(mass concentration). XRD patterns of sample d and e agreed well with the JCPDS (Joint Committee on Powder Diffraction Standards) PDF file 09-0169 for β-TCP,and no other crystalline phase was detected. However,samples with low gelatin concentration composed of a main phase of β-TCP and tiny phases of CPP (PDF 73-0440) and HA (PDF 09-0432). Samples without calcination all exhibited broad diffraction peaks and had the same apatitic structure of Ca-dHAP (Fig.2),but sample a,which was synthesized without gelatin,shows another monetite structure CaHPO4 (PDF 09-0080) except for the apatitic structure.
 | Fig.1 XRD patterns of the calcined powders (a) 0.00% gelatin; (b) 0.03% gelatin; (c) 0.05% gelatin; (d) 0.22% gelatin; (e) 0.42% gelatin |
 | Fig.2 XRD patterns of the as-prepared powders (a) 0.00%(mass concentration) gelatin,(b) 0.03% gelatin,(c) 0.05% gelatin,(d) 0.22% gelatin,(e) 0.42% gelatin |
FT-IR spectra of calcined powders showed in Fig.3 correspond well with the XRD patterns as shown in Fig.1. Bands representative of PO43- tetrahedral are visible at 556 cm-1 (O—P—O anti-symmetric bending ν4),607 cm-1 (O—P—O bending ν4),982 cm-1 (P—O bending ν1),and 1036—1136 cm-1(P—O stretching ν3). Bands at 760 cm-1,1164 cm-1,1210 cm-1 are attributed to P2O74- which comes from the decomposition of CaHPO4 in the as-prepared powders. These bands gradually vanished as the gelatin concentration increases above 0.22%(mass concentration),where pure β-TCP becomes the sole phase.
 | Fig.3 FT-IR spectra of the calcined powders (a) 0.00%(mass concentration) gelatin,(b) 0.03% gelatin,(c) 0.05% gelatin,(d) 0.22% gelatin,(e) 0.42% gelatin. |
Fig.4 illustrates the FT-IR spectra of the as-prepared powders. It clearly shows the presence of two characteristic ν4 PO43- bands at around 568 and 608 cm-1,and ν3 PO43- bands in the range of 9301200 cm-1 in all cases. The splitting of the PO43- ν3 and ν4 bands is the result of site-symmetry splitting of the modes as the environment of the PO43- groups are structurally ordered. Therefore,the featureless PO43- bands at 10201150 cm-1 suggest that the resulting crystals exhibit an amorphous structure,which is in agreement with the XRD results (Fig.2). A broad band in the high energy region from 3700 cm-1 to 2600 cm-1 and a band at about 1651 cm-1 can be assigned to the water molecule's stretching and bending modes,respectively. OH- groups of the apatite component of the as-prepared powders are indicated by a weak (but very sharp) band at 3570 cm-1 which can be assigned to the stretching mode appeared in all samples. Residual groups of nitrate (NO3-) were also discerned at 1381 cm-1. The spectra of carbonate ions (or carbonyl ions) appeared at 1460 and 1651 cm-1,whose intensity augmented as the amount of gelatin increased. In addition,the bands corresponding to HPO42- group are detected as a broad shoulder at about 876 cm-1. Additionally,amide A and amide B of gelatin exhibit bands at 30343265 cm-1,which overlap with the stretching modes of the water molecules,indicating that gelatin chemically bonded with the as-prepared powders.
 | Fig.4 FT-IR spectra of the as-prepared powders (a) 0.00%(mass concentration) gelatin; (b) 0.03% gelatin; (c) 0.05% gelatin; (d) 0.22% gelatin; (e) 0.42% gelatin |
The BET surface of as-prepared powders measured using N2 adsorption is given in Table 1. Powders prepared with the more amount of gelatin have the highest surface area (131.9 m2/g),while in verse with the slowest surface area (107.1 m2/g).
The mean crystallite size by using Scherrer equation for the (002) plane (as-prepared powder) and (300) plane (calcined powders) are shown in Table 1. The sample e has the smallest crystallites in the nanoscale size (16.8 nm) and sample a has the largest one. These results agree well with the specific surface area results (Table 1). Sample e has the smallest crystallites,since it was prepared with the more amount of gelatin. This means that gelatin can afford many nucleus sites for calcium and phosphorous ions to precipitate from the solution. It is well seen from the Table 1 that calcination can enhance the crystallite size and the calculated values are around 50 nm for the calcined powders,which is in the range of bone mineral crystallite size.
| Table 1 Crystallite sizes of the as-prepared powders and calcined powders |
Fig.5 shows a typical TEM image of as-prepared and calcined powders. The as-prepared powders were constituted of small agglomerated plate-like nano-sized crystals ( Fig.5A). The calcined powders composed of spherical particles which will merge together to form a botryoidal shape (Fig.5B). The morphology of the prepared powders is not changed greatly with the gelatin concentration. However,gelatin would change the nucleation process and subsequent phase transformation as is detailed in the following section.
 | Fig.5 TEM micrograph of the calcium phosphate powders (A) as-prepared nano-size powders;(B)calcined nano-size powders at 800 ℃ |
When being heated up to 800 ℃,samples contained gelatin suffered from three sharp weight losses phases (Fig.6). The first weight loss is the evaporation of the absorbed water between temperature 100 ℃300 ℃; the second weight loss happened at 300 ℃500 ℃,which is devoted to the carbon dioxide or water molecules coming from gelatin combustion; the third weight loss is the phase transformation of Ca-dHAP to β-TCP and CaHPO4 to Ca2P2O7. However,sample a without gelatin keeps a constant weight loss throughout the heat treatment process. The DTA curves are in good agreement with the TG curves. A broad endothermic peak appeared at about 200 ℃400℃ for both a and c,and two little sharp endothermic peaks emerged at about 300 ℃ for curve c,suggesting the beginning of combustion of gelatin to form carbonate dioxide or water molecules resulting in great weight loss. Furthermore,a little endothermic peak also appeared at 750 ℃ for sample c accompanying with a sharp weight loss around this temperature. And this weight loss was due to chemical reaction between HAP and Ca2P2O7 to form β-TCP which will be detailed below.
 | Fig.6 DTA/TG curves of as-prepared powders at a 10℃/min heating rate under static-air atmosphere (a) 0.00%(mass concentration) gelatin; (c) 0.05%gelatin |
Raynaud[21,22] et al. have found that the calcined powders composed of β-TCP and Ca2P2O7 as the Ca/P was below 1.5. They found that the precipitate nucleated from solution are composed of Ca9(HPO4)(PO4)5(OH) and CaHPO4. when the precipitate was calcined above 800 ℃,the precipitate will decompose according to the following equations:
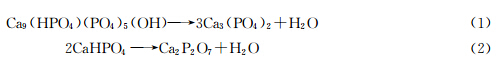
In our studies,the results agree well with the above mechanisms. However,pure β-TCP would become the sole phase as the gelatin concentration exceeds 0.22%(mass concentration). At first sight we may get an expression that gelatin can inhibit the formation of CaHPO4,and therefore the formation of Ca2P2O7. But our further studies indicated that the mechanism underlying the phase transformation is not so simple. When the as-prepared powders was calcined at 1000 ℃,sample d would decompose into two phases and sample e remain the sole β-TCP phase.
According to our results,the phase transformation during the heat treatment process may well be explained according to a mechanism proposed by Mortier et al[25].
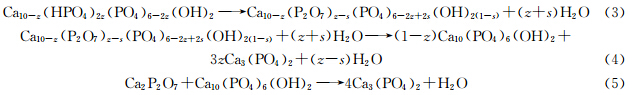
Gelatin can supply the nucleation site for the nucleation of Ca2+ and PO43- ions ( Scheme 1). The number of nucleation sites increased as the gelatin amount enhanced and more Ca-dHAP crystals would be formed with smaller crystallite size. Moreover,samples prepared with gelatin can absorb more water molecules when compared with the one without gelatin (Fig.4). According to equation (3)(5),the absorbed water molecules can react with Ca-dHAP to form HAP which will subsequently react with Ca2P2O7 to form β-TCP.
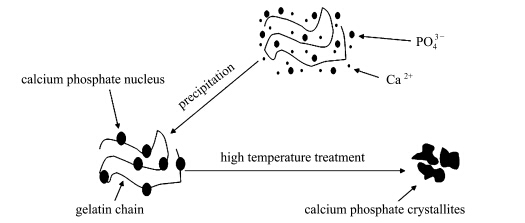
Scheme 1 The schematic nucleation process of calcium phosphate crystals in the gelatin contained solution and subsequent calcination process leading to the formation of β-TCP crystallites
3 ConclusionsPure β-TCP was prepared by adopting the biomineralization strategy and the precipitation method. The phosphate and carboxyl anion groups on the surface of gelatin could attract calcium ions by electrostatic force and act as nucleating sites for Ca-dHAP crystallization. Moreover,Ca-dHAP prepared with gelatin could absorb more water molecules and these water molecules would react with Ca-dHAP to form HAP,which will subsequently react with Ca2P2O7 to form β-TCP. The gelatin induced pure β-TCP is proposed to find application in the high-performance liquid chromatography (HPLC) for separating biopolymers,as well as in the bone defect substitution or repair in the form of cement or biphasic calcium phosphate composites mixed with HA.
| [1] | Dorozhkin SV, Epple M. Biological and medical significance of calcium phosphates[J]. Angewandte Chemie-International Edition, 2002, 41(17): 3130-3146. |
| [2] | Yuan H, Groot K. In: Reis R L, Weiner S, editors. Calcium Phosphate Biomaterials: An Overview[M]. Netherlands: Springer, 2005. 37-57. |
| [3] | Hench L L. Bioceramics - from Concept to Clinic[J]. Journal of the American Ceramic Society, 1991, 74(7): 1487-1510. |
| [4] | Stankewich C J, Swiontkowski M F, Tencer A F, et al. Augmentation of femoral neck fracture fixation with an injectable calcium-phosphate bone mineral cement[J]. Journal of Orthopaedic Research, 1996,14(5): 786-793. |
| [5] | Ducheyne P, Cuckler J M. Bioactive ceramic prosthetic coatings[J]. Clinical Orthopaedics and Related Research, 1992, 276:102-114. |
| [6] | Grundel R E, Chapman M W, Yee T, Moore D C. Autogenic bone-marrow and porous biphasic calcium phosphate ceramic for segmental bone defects in the canine ulna[J]. Clinical Orthopaedics and Related Research, 1991, 266: 244-258. |
| [7] | Lu J, Descamps M, Dejou J, et al. The biodegradation mechanism of calcium phosphate biomaterials in bone[J]. Journal of Biomedical Materials Research, 2002, 63(4): 408-412. |
| [8] | Gauthier O, Müller R, von Stechow D, et al. In vivo bone regeneration with injectable calcium phosphate biomaterial: A three-dimensional micro-computed tomographic, biomechanical and SEM study[J]. Biomaterials, 2005,26(27): 5444-5453. |
| [9] | Kivrak N, Tas A C. Synthesis of calcium hydroxyapatite-tricalcium phosphate (HA-TCP) composite bioceramic powders and their sintering behavior[J]. Journal of the American Ceramic Society, 1998,81(9): 2245-2252. |
| [10] | Murugan R, Ramakrishna S. Crystallographic study of hydroxyapatite bioceramics derived from various sources[J]. Crystal Growth & Design, 2004, 5(1): 111-112. |
| [11] | Hench L L. Bioceramics[J]. Journal of the American Ceramic Society, 1998, 81(7): 1705-1728. |
| [12] | Rejda B V, Peelen J G J, Groot K D. Tricalcium phosphate as a bone substitution[J]. Journal of Bioengineering, 1977, 1(2): 93-97. |
| [13] | Braye F, Irigaray J L, Jallot E, et al. Resorption kinetics of osseous substitute: Natural coral and synthetic hydroxyapatite[J]. Biomaterials, 1996,17(13): 1345-1350. |
| [14] | Bow J S, Liou S C, Chen S Y. Structural characterization of room-temperature synthesized nano-sized [beta]-tricalcium phosphate[J]. Biomaterials, 2004, 25(16): 3155-3061. |
| [15] | Okazaki M, Sato M. Computer graphics of hydroxyapatite and [beta]-tricalcium phosphate[J]. Biomaterials, 1990, 11(8): 573-578. |
| [16] | Pang Y X, Bao X. Influence of temperature, ripening time and calcination on the morphology and crystallinity of hydroxyapatite nanoparticles[J]. Journal of the European Ceramic Society, 2003, 23(10): 1697-1704. |
| [17] | Mostafa N Y. Characterization, thermal stability and sintering of hydroxyapatite powders prepared by different routes[J]. Materials Chemistry and Physics, 2005, 94(2-3): 333-341. |
| [18] | Liou S C, Chen S Y. Transformation mechanism of different chemically precipitated apatitic precursors into [beta]-tricalcium phosphate upon calcination[J]. Biomaterials, 2002, 23(23): 4541-4547. |
| [19] | Cuneyt Tas A, Korkusuz F, Timucin M, et al. An investigation of the chemical synthesis and high-temperature sintering behaviour of calcium hydroxyapatite (HA) and tricalcium phosphate (TCP) bioceramics[J]. Journal of Materials Science: Materials in Medicine, 1997,8(2): 91-96. |
| [20] | Ishikawa K, Ducheyne P, Radin S. Determination of the Ca/P ratio in calcium-deficient hydroxyapatite using X-ray diffraction analysis[J]. Journal of Materials Science: Materials in Medicine, 1993,4(2): 165-168. |
| [21] | Raynaud S, Champion E, Bernache A D, et al. Calcium phosphate apatites with variable Ca/P atomic ratio I.Synthesis, characterisation and thermal stability of powders[J]. Biomaterials, 2002,23(4): 1065-1072. |
| [22] | Raynaud S, Champion E, Bernache A D. Calcium phosphate apatites with variable Ca/P atomic ratio II. Calcination and sintering[J]. Biomaterials, 2002, 23(4): 1073-1080. |
| [23] | Kikuchi M, Itoh S, Ichinose S, et al. Self-organization mechanism in a bone-like hydroxyapatite/collagen nanocomposite synthesized in vitro and its biological reaction in vivo[J]. Biomaterials, 2001,22(13): 1705-1711. |
| [24] | Hartgerink J D, Beniash E, Stupp S I. Self-assembly and mineralization of peptide-amphiphile nanofibers[J]. Science, 2001,294(5547): 1684-1688. |
| [25] | Mortier A, Lemaitre J, Rouxhet P G. Temperature-programmed characterization of synthetic calcium-deficient phosphate apatites[J]. Thermochimica Acta, 1989,143: 265-282. |