




主体成膜树脂是光刻胶最主要的成分之一,它的性能好坏对光刻胶性能有决定性的影响.它是光刻胶的骨架,是光刻胶的基础材料.为了提高光刻分辨率,光刻技术的曝光波长已经从紫外g线的436 nm、i线的365 nm,缩短到KrF准分子激光的248 nm,再到ArF准分子激光的193 nm[ 1 ].相应地,光刻胶所需的主体成膜树脂也从聚乙烯醇肉桂酸酯类、环化橡胶类发展到线性酚醛树脂类,再发展到对羟基苯乙烯类和不含芳环的多脂 环体系类[ 2,3,4,5 ].
当曝光波长缩短到248 nm后,原有的光刻胶的成膜树脂如线性酚醛树脂等因含有过多的苯环而在248 nm处有强烈的光学吸收(其光学密度大于1 μm-1)[ 6 ],就不能继续用作248 nm光刻胶的成膜树脂.出于对光透明度的考虑,最早用于248 nm光刻胶成膜树脂的材料是聚甲基丙烯酸酯,因其不含芳环,在248 nm处高度透明,分辨率也高,但它主要是通过主链断裂来成像的,所需曝光能量高,曝光灵敏度低,限制了它的使用.
248 nm光刻胶,普遍采用化学增幅技术[ 2 ],其成膜树脂的开发研制经历了相当长的时间.从20世纪70年代后期开始,人们经过长期的尝试,最终将研究的重点集中在了聚羟基苯乙烯及其衍生物上[ 7,8,9,10,11 ].由于聚羟基苯乙烯中含有大量的苯环,因此它的抗干蚀刻能力不是问题;通过对其在碱溶液中溶解性能、热稳定性等方面的详细研究,结果表明,聚对羟基苯乙烯在溶解性和热稳定性等方面比邻位和间位的聚羟基苯乙烯要优越,而且纯的聚对羟基苯乙烯在248 nm处是光学透明的(光学密度为0.22 μm-1,即1 μm厚度的胶膜,其吸光度为0.22)[ 12,13 ],而酚羟基被特丁氧酰基保护的聚对羟基苯乙烯在248 nm处更透明(光学密度仅为0.1 μm-1),因此人们最终选择了聚对羟基苯乙烯及其衍生物作为248 nm光刻胶的成膜树脂.目前商品化的248 nm光刻胶大都采用部分保护的对羟基苯乙烯及其与其它单体的共聚物作为成膜树脂,一些已经公开的聚合物具有如图1中式Ⅰ—Ⅴ的结构[ 14,15,16,17 ].
 | 图1 一些含有对羟基苯乙烯单体结构的成膜树脂 Some matrix resins containing monomer p-hydroxystyrene structure |
本文制备了一种具有图1结构Ⅵ的聚合物(即聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯-共-对特丁氧酰氧基苯乙烯),并探讨了它的紫外吸收性能、热性能、抗干蚀刻性能、通过热解和酸解脱除保护基的性能,测试了它的酸解留膜率曲线.结果表明,如果分子量和酚羟基上的特丁氧酰保护基脱除比例(即脱保率)合适,它有可能被用作248 nm光刻胶的成膜树脂. 1 实验部分 1.1 仪器与试剂
红外光谱(FT-IR)分析采用EQUINQX 55型红外光谱仪(德国Bruker公司),KBr压片;1H核磁共振分析采用Mercury VX-300 型核磁共振仪(美国Varian公司),四甲基硅烷(TMS)为内标,300 MHz;热性能测试采用DSC-7型、TGA-7型热分析仪(美国Perkin Elmer公司),N2气氛,10 ℃/min升温速率;聚合物分子量采用Waters 2695型凝胶渗透色谱仪(Waters 2695 GPC)(美国Waters公司)测试,四氢呋喃(THF)为流动相,流速1.00 mL/min,柱温23 ℃;抗干蚀刻能力实验采用ME-3A型多功能磁增强反应离子蚀刻机(中科院微电子中心),蚀刻参数为:源:50 W、5 min,100 W、3 min;CHF3:20 ccm;SF6:80 ccm;O2:5 ccm;紫外吸收光谱测试采用UV-2550型紫外可见光分光光度计(SHIMADZU公司),测定时以空白试片为参比,扫描步速为1 nm;胶膜的厚度和均匀性采用Dektak Ⅱ型探针轮廓仪Profilometer(美国VEECO公司)测试,测试精度为1×10-10 m(即0.1 nm).
N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMAc)、乳酸乙酯、四氢呋喃(THF)、二氧六环、偶氮二异丁腈(AIBN)、三氟甲基磺酸等为分析纯试剂,乙二醇单甲醚乙酸酯为化学纯试剂,三氟甲基磺酸铵为自制试剂. 1.2 材料制备与性能表征 1.2.1 单体及前驱体聚合物聚特丁氧酰氧基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯的制备 1.2.1 .1 单体的制备
单体对特丁氧酰氧基苯乙烯参考文献[ 18,19,20 ]制备.FT-IR:1760 cm-1(νCO),1630 cm-1(νCC),1370 cm-1和1394 cm-1(特丁基的特征吸收:低波数强度约为高波数的两倍),990 cm-1(δC—H);1HNMR(CDCl3):1.487(9H,s,—C(CH3)3),5.129—5.637(2H,m,CH2),6.552—6.647(1H,m,CH),7.020—7.323(4H,m,—C6H4—).
单体N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯参考文献[ 21 ]制备.m.p.136—138 ℃;FT-IR:3070、3010 cm-1(νC—H),2958、2884 cm-1(νC—H),1760、1734 cm-1(νCO),1635 cm-1(νCC);1HNMR(CD3COCD3):1.630—1.725 (2H,m,—CH2—),1.993(3H,s,—CH3),3.379、3.506(4H,两个单峰,—CH—),5.963(2H,CH),6.156、6.278(2H,两个单峰,CH2). 1.2.1 .2前驱体聚合物的制备
聚合瓶中加入4.94 g (0.020 mol) N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯和40 mL DMF,溶解,再加入4.40 g (0.020 mol)对特丁氧酰氧基苯乙烯,通10 min 高纯氩气(Ar)除氧,然后加入0.070 g的引发剂AIBN,密封聚合瓶,抽真空除氧,然后通Ar,反复三次,放入65 ℃恒温水浴中,聚合24 h.取出冷至室温,缓慢加入至大量去离子水中,有白色沉淀出现,抽滤,用去离子水洗涤,65 ℃真空干燥至恒重,得9.1 g前驱体聚合物,聚合收率97.4%.其提纯方法为:将干燥后的聚合物重新溶于DMF中,然后滴入去离子水中,沉淀,抽滤,真空烘干,反复三次.所得前驱体聚合物的数均分子量Mn=1.59×104,重均分子量Mw=2.43×104,分子量分布系数为1.53.
要制备其它分子量的前驱体聚合物,可采用类似的聚合工艺和步骤,仅需改变加入引发剂AIBN的量即可. 1.2.2前驱体聚合物完全脱除酚羟基保护基,制备聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯
参考文献[ 18 ],溶解5.0 g(0.011 mol)上述前驱体聚合物于30 mL二氯甲烷中,将0.30 g(0.0026 mol)三氟甲基磺酸溶于10 mL二氯甲烷中,然后将三氟甲基磺酸的二氯甲烷溶液逐滴滴入前驱体聚合物的二氯甲烷溶液中,有气体逸出,滴加完毕,搅拌,待无气体逸出(即酚羟基上的特丁氧酰保护基完全脱除)时,停止搅拌,将体系滴入石油醚中,有沉淀析出,抽滤,真空烘干,即得聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯.其提纯方法为:将干燥后的聚合物重新溶于DMF中,然后滴入去离子水中,沉淀,抽滤,真空烘干,反复三次.
当前驱体聚合物的Mn=1.59×104和Mw=2.43×104时,按上述步骤,完全脱除保护基后,所得聚合物的Mn=1.18×104和Mw=1.90×104,分子量分布系数为1.61.
其它分子量的聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯,可通过不同分子量的前驱体聚合物按上述工艺和步骤,完全脱除酚羟基上的保护基而得到. 1.2.3目标聚合物聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯-共-对特丁氧酰氧基苯乙烯(Ⅵ)的制备
将5.0 g上述前驱体聚合物放入一定温度(通常为150—180 ℃)的烘箱中,加热一定时间,取出,冷至室温,即得目标聚合物;其提纯方法为:将所得聚合物重新溶于DMF中,然后滴入去离子水中,沉淀,抽滤,真空烘干,反复3次.
当前驱体聚合物的Mn=1.59×104和Mw=2.43×104时,按上述步骤,在温度155 ℃烘箱中加热2—3 min,可得到脱保率约为60%的目标聚合物,其Mn=1.38×104,Mw=2.15×104,分子量分布系数为1.56.
其它分子量的目标聚合物可通过不同分子量的前驱体聚合物按上述脱保工艺和步骤,部分脱除酚羟基上的保护基而得到;其脱保率可通过改变上述工艺中的脱保加热温度和/或加热时间而改变. 1.2.4目标聚合物成膜性能、亲油性、膜厚度、紫外吸收光谱和酸解留膜率的测试
将上述制得的目标聚合物0.30 g溶于2.0 g 乙二醇甲醚乙酸酯与乳酸乙酯的混合溶剂(1∶1体积比)中,得聚合物溶液,用0.25 μm超滤膜过滤,然后用旋转涂布机涂在高纯石英片或单晶硅片上,转速3000—7000 r/min,最后在90 ℃烘盘上烘5 min,在光学显微镜下观察聚合物膜厚是否有裂纹和起皮(亲油性)、是否有不溶的固体颗粒(溶解性)等,用紫外可见分光光度计测其紫外吸收光谱.
当用探针轮廓仪测试聚合物膜的厚度和均匀性时,先用丙酮脱脂棉球将单晶硅片表面某一边一定宽度的聚合物膜彻底擦拭掉,然后再90 ℃烘5 min,再进行测试.
酸解留膜率测试:向上述聚合物溶液中加入1%(质量分数)的三氟甲基磺酸铵,溶解,用0.25 μm超滤膜过滤,然后旋涂于单晶硅片上,在不同温度烘盘上烘3 min,冷却,用探针轮廓仪测试胶膜的厚度,记为f1;然后将其放入浓度为2.38%(质量分数)的氢氧化四甲基铵溶液中3 min,取出,用去离子水冲洗干净,再在90 ℃烘盘上烘干,再测试胶膜的厚度,记为f2,酸解留膜率即为f2/f1×100%. 2 结果与讨论 2.1 前驱体聚合物及目标聚合物的制备与FT-IR谱
单体对特丁氧酰氧基苯乙烯与N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯通过自由基共聚合,可制备前驱体聚合物聚特丁氧酰氧基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯.该前驱体聚合物酚羟基上的特丁氧酰基保护基,在酸性和/或加热条件下,都可以被脱除[ 18 ],但通过控制加热时间或温度更容易控制脱保率,因此本研究采用在加热条件下部分脱除酚羟基上的特丁氧酰基保护基,得到目标聚合物.上述聚合和脱保过程可用式(1)表示.前驱体聚合物之所以不能直接使用,是因为其酚羟基的保护基完全脱除前后形成的聚合物胶膜有过大的体积收缩率,导致胶膜容易开裂[ 2 ].
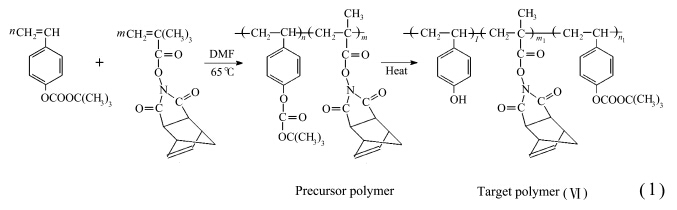
前驱体聚合物(Mn=1.59×104,Mw=2.43×104)的FT-IR谱如图2中的曲线1所示,2981 cm-1、2942 cm-1、2870 cm-1等为甲基或者碳链上的νC—H吸收,1734 cm-1为甲基丙烯酸酯羰基的νCO吸收,1668 cm-1、1778 cm-1、1807 cm-1为酰亚胺羰基的νCO吸收,而特丁氧酰基的羰基吸收被掩盖,1612 cm-1、1508 cm-1、1456 cm-1为苯环骨架振动的ν<sub>的吸收,1369 cm-1和1390 cm-1为特丁基的特征吸收,1275 cm-1、1256 cm-1、1216 cm-1、1146 cm-1为νC—O—C的吸收,835 cm-1为对位取代苯环的δC—H吸收,与其结构相符.目标聚合物(Mn=1.38×104,Mw=2.15×104,约为60%脱保率)的FT-IR如图2中的曲线2所示,由于酚羟基的生成,图中明显出现了最大吸收位置在3500 cm-1左右的宽带吸收峰.
 | 图2 前驱体聚合物(曲线1,Mn=1.59×104,Mw=2.43×104)和目标聚合物 (曲线2,Mn=1.38×104,Mw=2.15×104,约60%脱保率)的FT-IR谱 FT-IR spectra of precursor (curve 1,Mn=1.59×104,Mw=2.43×104) and target (curve 2,Mn=1.38×104,Mw=2.15×104,about 60% removal ratio of the protective group) polymers |
图3中曲线2和3分别为前驱体聚合物(Mn=1.59×104,Mw=2.43×104)的TGA和DSC曲线,可以看出,它的Tg大约为180 ℃,第一阶段的起始分解温度在150 ℃左右,第二阶段的起始分解温度在270 ℃左右.到193 ℃第一阶段分解结束时,失重约21.4%.若该聚合物是严格的1∶1(摩尔比)的共聚物,当全部酚羟基上的保护基失去后(即脱保率为100%),失重正好为21.4%.由此可以推断,聚合时,若投料比是1∶1,通过自由基共聚合得到的聚合物应该是1∶1的共聚物.
 | 图3 前驱体聚合物(曲线2和3,Mn=1.59×104,Mw=2.43×104)和目标聚合物 (曲线1,Mn=1.38×104,Mw=2.15×104,约60%脱保率)的TGA(曲线2和1)和DSC曲线(曲线3) TGA (curves 2 and 1) and DSC (curve 3) curves of precursor (curves 2 and 3,Mn=1.59×104,Mw=2.43×104) and target (curve 1,Mn=1.38×104,Mw=2.15×104,about 60% removal ratio of the protective group) polymers |
实验表明,通过同一前驱体聚合物的热分解而得到的目标聚合物,具有与相应前驱体聚合物类似的热性能,只是随着保护基含量的不同,其第一阶段分解结束时,失重的百分含量有所变化(即可以通过热重分析,来测定脱保后聚合物中酚羟基的含量).例如,当我们将运用文献[ 22 ]的方法测得的酚羟基含量约为40%(即脱保率约为60%)的目标聚合物(Mn=1.38×104,Mw=2.15×104)进行TGA分析,在第一阶段分解结束时,发现其失重约为12.80%(图3的曲线1),这与理论计算得到的失重比率(12.84%)也是相符的.此外,当制备不同脱保率的目标聚合物时,发现升高加热温度或延长加热时间,都可以提高脱保率,而升高加热温度,还可以提高脱保速率.
进一步实验表明,目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)具有良好的有机溶剂溶解性能,可溶于DMF、N,N-二甲基乙酰胺(DMAc)、THF、二氧六环、乳酸乙酯、丙二醇甲醚乙酸酯等多种有机溶剂,形成的溶液经旋涂并烘干后,可在单晶硅等衬底表面形成聚合物薄膜,该薄膜可以很好的附着于衬底上.图4为用探针轮廓仪测得的其中两个薄膜的厚度曲线(即每个图中间位置有台阶的曲线,台阶左边为探针在硅片上的测试线,台阶右边为在聚合物薄膜上的测试线).可以看出,薄膜的厚度(即图中的VERT值)分别为499 nm(0.499 μm)和469.8 nm(0.4698 μm),在聚合物薄膜上的测试线几乎是平直的,且多个样品、多次测试均能得到类似的结果,这表明薄膜厚度具有很好的均匀性.
 | 图4 由探针轮廓仪测得的目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)薄膜的厚度曲线 Thickness curve of target polymer (Mn=1.38×104,Mw=2.15×104, about 60% removal ratio of the protective group) film,measured by a probe profilometer |
光刻胶的抗干蚀刻能力与光刻胶中成膜树脂的结构与种类密切相关,其评价标准通常有两种,第一种是光刻胶的抗干蚀刻能力与其成膜树脂中所含的有效碳含量有关[ 23 ],即:
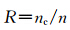
第二种评价标准是光刻胶的抗干蚀刻能力与其成膜树脂的蚀刻速度有关[ 24 ],即:
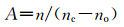
本文采用第一种评价标准,前驱体聚合物的有效碳含量R=41.27%.当保护基部分脱除(即得目标聚合物)或全部脱除后,有效碳含量会进一步升高.抗干蚀刻能力实验也表明,当采用如前1.1节所述的蚀刻参数(该蚀刻参数常用于对以线性酚醛树脂为成膜树脂的紫外光刻胶(如AZ-1500光刻胶)的刻蚀)进行蚀刻后,发现目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)薄膜表面并未发生明显的变化,这表明该聚合物的抗干蚀刻能力与线性酚醛树脂的相当.
目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)的紫外吸收曲线如图5所示,可以看出,在248 nm时,其光学密度为0.212 μm-1.而文献报道[ 25 ],对于248 nm光刻胶,当成膜树脂的光学密度小于0.45 μm-1时,即可满足要求.因此,目标聚合物的紫外透光性可以满足248 nm光刻胶的需要.
 | 图5 目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)的紫外吸收曲线 UV-Vis spectrum of target polymer (Mn=1.38×104,Mw=2.15×104, about 60% removal ratio of the protective group) |
用于光刻胶成膜树脂的分子量通常在数千至数万之间[ 2,23 ].在此范围内实验发现,目标共聚物的有机溶剂溶解性能、成膜性能、紫外光透过性能并没有明显变化;且在此分子量范围内,目标聚合物的脱保率对这些性能也没有明显的影响;但热性能随分子量的变化有所变化,分子量越大,玻璃化温度Tg也越高,耐热性越好. 2.3 目标聚合物的酸致脱保性能和酸解留膜率
在化学增幅型光刻胶的曝光过程中,曝光区域的光致酸发生剂会产生酸,这些酸可以进一步催化酸增殖剂产生更多的酸.若曝光区域光刻胶的成膜树脂采用特丁氧酰基或缩醛基等作为其酚羟基的保护基,那么所有产生的酸都可以催化脱除这些保护基,而随后的曝光后烘工艺还可以使这一过程进行得更为彻底[ 2 ],这种性能即为成膜树脂的酸致脱保性能.本文中制备的目标共聚物,实验证实可以用三氟甲基磺酸完全脱除其酚羟基上的特定氧酰保护基,表明目标聚合物具备酸致脱保性质,这是目标聚合物可用于248 nm光刻胶成膜树脂的重要性能之一.目标聚合物(Mn=1.38×104,Mw=2.15×104,约60%脱保率)及其完全脱除特丁氧酰保护基后的FT-IR如图6所示,可以看出,在完全脱保后的红外吸收曲线2中,未发现酚羟基的特丁氧酰氧保护基在1371 cm-1、1395 cm-1(叔丁基的δC—H吸收)及1750 cm-1(羰基的νCO吸收)附近的特征吸收峰,但同时出现了更强的酚羟基吸收(3100—3600 cm-1,最大吸收在3420 cm-1左右).
文献[ 15 ]报道,三氟甲基磺酸铵在一定温度下是稳定的,但当加热到更高温度时会发生分解,并产生三氟甲基磺酸,因此可以用三氟甲基磺酸铵热解产酸代替实际光刻胶中的光致产酸,进而对可酸致脱保的聚合物进行留膜率测试.图7为目标聚合物(约60%脱保率,Mn=1.38×104,Mw=2.15×104)的酸解留膜率曲线,可以看出,在烘烤温度约为120 ℃时,曲线上有明显的拐点出现,表明此时目标聚合物经酸解部分脱保,开始溶于氢氧化四甲基铵水溶液,当烘烤温度升至140 ℃时,留膜率为零,表明此时目标聚合物能可完全溶于氢氧化四甲基铵水溶液.
 | 图6 目标聚合物(曲线1:Mn=1.38×104,Mw=2.15×104,约60%脱保率)及其完全脱除保护基后(曲线2)的FT-IR谱 FT-IR spectra of target polymer (Mn=1.38×104,Mw=2.15×104, about 60% removal ratio of the protective group) before and after totally removing protective group |
 | 图7 目标聚合物(Mn=1.38×104,Mw=2.15×104)的酸解留膜率曲线 Remaining thickness ratio of the target polymer (Mn=1.38×104,Mw=2.15×104) after acidolysis |
进一步实验表明,当目标聚合物的分子量太小(如1000以下)时,无论目标聚合物中酚羟基保护基的含量是多少,它都易溶于氢氧化四甲基铵水溶液,因此在测试酸解留膜率曲线时,没有明显的拐点出现.若这样分子量的目标聚合物被用作光刻胶的成膜树脂,推测其在曝光显影时,将不会有成像反差,即无法成像.而当目标聚合物的分子量在数千至数万之间时,若其酚羟基的脱保率小于约70%,目标聚合物就难溶于氢氧化四甲基铵水溶液.之后,随着脱保率的增大,目标聚合物在碱性水溶液中逐渐开始溶解,因此在其酸解留膜率曲线上出现了明显的拐点.这样分子量的目标聚合物若被用作光刻胶的成膜树脂,推测其在曝光显影时,将会有明显的成像反差.因此,分子量在数千至数万的目标聚合物才适合用作光刻胶的成膜树脂.
此外,目标聚合物上酚羟基的详细保护比例,以及由这些目标聚合物作为成膜树脂配制而成的光刻胶的配方、详细光刻工艺和光刻分辨率等问题,作者正在进一步研究中. 3 结论
(1)通过自由基共聚合,制备了前驱体共聚物聚对特丁氧酰氧基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯,该共聚物可以在加热条件下脱除酚羟基上的部分保护基,得到目标共聚物聚对羟基苯乙烯-共-N-羟基-5-降冰片烯-2,3-二甲酰亚胺甲基丙烯酸酯-共-对特丁氧酰氧基苯乙烯.
(2)具有合适分子量的目标共聚物,其溶解性、热性能、成膜性能、抗干蚀刻性能、紫外光透过性能等可以初步满足248 nm光刻胶成膜树脂的需要.
(3)从目标共聚物的酸致脱保性能和酸解留膜率曲线可以推测,当其分子量和脱保率均合适时,目标聚合物有可能满足光刻胶的曝光显影成像功能.
| [1] | John H B. Optical lithography-thirty years and three orders of magnitude [J]. Proceedings of SPIE, 1997, 3049: 14-27. |
| [2] | Ito H. Chemically amplified resist: past, present and future[J]. Proceedings of SPIE, 1999, 3678: 2-12. |
| [3] | 穆启道,曹立新. 光刻技术的发展与光刻胶的应用[J]. 集成电路应用,2003,6:69-75. Mu Q D, Cao L X. Development of photolithographic technology and application of photoresist [J]. Applicatios of IC, 2003, 6: 69-75. |
| [4] | 吴雄杰,裘霞敏. 我国化学电子品产业的发展状况及前景[J]. 浙江化工,2003,34(6):25-27. Wu X J, Qiu X M. Development and prospect of electronic chemicals industry in China [J]. Zhejiang Chemical Industry, 2003, 34(6): 25-27. |
| [5] | 夏伟如,章 舒,夏 敏,等. 正性抗蚀剂用酚醛树脂的制备[J]. 化工时刊,2002,4:21-24. Xia W R, Zhang S, Xia M, et al. Preparation of cresol-formaldehyde resins used for making positive photoresist [J]. Chemical Industry Times, 2002, 4: 21-24. |
| [6] | Strurtevant J, Conley W E. Photosensitization in dyed and undyed APEXE DUV resist [J]. Proceedings of SPIE, 1996, 2724: 273-279. |
| [7] | Thackery J W, Orsula G W. Deep UV photoresist for 248 nm excimer laser photolithography [J]. Proceedings of SPIE, 1989, 1086: 34-44. |
| [8] | Hayashi K I, Kikuchi H. Charateristics of new KrF excimer laser resist [J]. Proceedings of SPIE, 1990, 1262: 468-475. |
| [9] | Harry F. Evaluation of resist materials for KrF excimer laser lithography [J]. Proceedings of SPIE, 1990, 1262: 331-343. |
| [10] | Robert D A, Quan P L, Gregory M W, et al. New Chemistry in the design of chemistry amplified positive resists [J]. Proceedings of SPIE, 1993, 1925: 246-256. |
| [11] | Willson C G, Dammel R A, Reiser A, et al. Photoresist materials: a historical perspective [J]. Proceedings of SPIE, 1997, 3049: 28-41. |
| [12] | Ito H, Willson C G. Positive/negative mid UV resist with high thermal stability [J]. Proceedings of SPIE, 1987, 771: 24-30. |
| [13] | Ito H, Pederson L A. Sensitive electron beam resist systems based on acid-catalyzed deprotection [J]. Proceedings of SPIE, 1989, 1086: 11-17. |
| [14] | Colley W, Breyta G, Brunsvold B, et al. The lithographic performance of an environmentally stable chemically amplified photoresist (ESCAP) [J]. Proceedings of SPIE, 1996, 2724: 34-60. |
| [15] | Tanabe T, Kobayashi Y, Tsuji A. PED stabilized chemically amplified photoresist [J]. Proceedings of SPIE, 1996, 2724: 61-69. |
| [16] | Choi S J, Jung S Y, Kim C H, et al. Design and properties of new deep-UV positive photoresist [J]. Proceedings of SPIE, 1996, 2724: 323-331. |
| [17] | 刘建国,郑家燊,李 平. 聚羟基苯乙烯在光致抗蚀剂中的应用及其合成[J]. 感光科学与光化学,2006,24(1):67-74. Liu J G, Zheng J S, Li P. Application and syntheses of polyhydroxystyrene in deep UV photoresist [J]. Photographic Science and Photochemistry, 2006, 24(1): 67-74. |
| [18] | Frechet J M J, Eichler E. Poly (p-tert-butoxycarbonyloxystyrene): a convenient precursor to p-hydroxystrene resins [J]. Polymer, 1983, 24(8): 995-1000. |
| [19] | 刘建国,李 平,刘和平,等. 对特丁氧酰氧基苯乙烯的制备新方法[J]. 应用化学,2008, 25(4):424-428. Liu J G, Li P, Liu H P, et al. A novel synthesis of p-tert-butoxycarbonyloxystyrene [J]. Chinese Journal of Applied Chemistry, 2008, 25(4): 424-428. |
| [20] | 刘建国,李 平,李 萍,等. 聚对羟基苯乙烯单体的中间体—对特丁氧酰氧基苯乙烯合成方法的改进[J]. 应用化学,2007,24(3):361-364. Liu J G, Li P, Li P, et al. Synthetic improment of p-tert-butoxycarbonyloxystyrene -intermediate of monomer of poly(p-hydroxystyrene) [J]. Chinese Journal of Applied Chemistry, 2007, 24(3): 361-364. |
| [21] | 陆承勋,王东法,冯新德,等. N-丙烯酰氧-5-降冰片烯-2,3-双甲酰亚胺的合成及聚合[J]. 科学通报,1981,26(4):214-216. Lu C X, Wang D F, Feng X D, et al. Synthesis and polymerization of N-hydroxy-5-norbornene-2,3-dicarboximido methacrylate [J]. Chinese Science Bulletin, 1981, 26(4): 214-216. |
| [22] | 复旦大学高分子科学系高分子科学研究所 编著. 高分子实验技术[M]. 上海:复旦大学出版社,1996. 318. Polymer Research Institute, Department of Polymer Science, Fudan University. Polymer Experiment Technology[M]. Shanghai: Fudan University Press, 1996. 318. |
| [23] | 王春伟,李 弘,朱晓夏. 化学放大光刻胶高分子材料研究进展[J]. 高分子学报,2005,2:70-80. Wang C W, Li H, Zhu X X. Progress in the research of chemical amplified photoresist [J]. Polymer Bulletin, 2005, 2: 70-80. |
| [24] | 武 玲,余尚先. 正性光致抗蚀剂主要成膜树脂—酚醛树脂[J]. 感光材料,1994,4:3-8, 43. Wu L, Yu S X. Main matrix resin for positive photoresist-phenolic resins [J]. Image Matererials, 1994, 4: 3-8, 43. |
| [25] | Conley W. Consideration in the development of deep UV photoresist material & processes [J]. Proceedings of SPIE, 1995, 2438: 40-52. |