




2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, P. R. China
化石燃料的使用导致了严重的环境污染和温室效应[1],因而寻找新的、对环境友善的能源势在必行[2]。通过电解水获取氢气和氧气是理想的途径之一,但水氧化半反应是实现水分解的瓶颈[3, 4]。至今已有许多此类反应的过渡金属催化剂被报道,主要是基于钌[5, 6]、铱[6, 7]、锰[8]、钴[9, 10, 11]、镍[12, 13, 14]和铁[15]的配合物。但寻找廉价易得、活性高并且稳定性好的催化剂仍面临诸多挑战。
铜是一种来源广泛的非贵金属,而且铜对生物体毒性较小。近几年,基于铜构建的水氧化催化剂开始受到关注[16]。自2012年Mayer等人首次报道了铜联吡啶化合物在碱性条件下催化电解水产氧[17]之后,铜盐与四聚甘氨酸原位生成的配合物[18],以及在碳酸钠溶液中的铜盐[19]均被证明有催化作用。2014年,杜平武等人通过在碱性溶液中电解铜的二甲氨基吡啶化合物获得具有电催化产氧活性的CuO[20]。2015年,孙立成等又报道了铜盐在硼酸缓冲体系中电沉积产生具有催化产氧作用的Cu-Bi薄膜[21]。
研究表明商业CuO并无催化活性[20],而使用Cu盐原位制备的CuO活性和稳定性都低于由Cu配合物制备的CuO[19, 20]。为获得廉价易得的产氧催化剂,我们使用商业化试剂N,N,N′,N′-四羟乙基乙二胺(THEED)和无水硫酸铜制备得到配合物[Cu(THEED)(H2O)]SO4。它在碱性水溶液中具有良好的溶解性,可以有效地避免Cu(OH)2沉淀析出。本文采用[Cu(THEED)(H2O)]SO4作为前驱体,在配合物晶体结构确定的基础上,通过阳极表面电沉积方法原位得到具有较高稳定性和电催化活性的CuO,同时实现了电催化的水氧化。
1 实验部分 1.1 仪器与试剂仪器: CHI660C电化学工作站(上海辰华仪器公司);APEXII型FT-ICR电喷雾质谱仪;EURO EA元素分析仪;Hitachi UV-3010紫外-可见吸收光谱仪;Hitachi S-4800扫描电子显微镜;D8 focus X射线衍射仪;Rigaku R-AXIS RAPID IP X-ray单晶衍射仪;X射线光电子能谱仪(PHI Quantera SXM);Ocean Optics optical probe氧气监测探针。
试剂:N,N,N′,N′-四羟乙基乙二胺(Aldrich公司);无水CuSO4、NaOAc、NaOH均购于北京化工厂,用于化学反应和测试时未经进一步处理直接使用。所有用于实验的水均是Milli-Q超纯水(> 18 MΩ)。
1.2 [Cu(THEED)(H2O)]SO4配合物的合成将5.0 mmol的无水硫酸铜溶于10 mL乙醇和水(体积比1∶1)中,在搅拌下加入5.0 mmol的THEED。室温搅拌3 h后,旋干溶剂得粗产物。于乙醇中重结晶得到纯品,产率为90%。ESI-MS(H2O,m/z):396.0602 [M-H2O + H]+,149.5506[M-H2O-SO4]2+。Anal.Calcd. for[Cu(THEED)(H2O)]SO4:C,29.02;H,6.33;N,6.77 %. Found: C,27.91;H,6.49;N,6.49 %。
1.3 电化学测试 1.3.1 循环伏安测试用玻璃碳电极(面积0.07 cm2)或ITO导电玻璃(25 mm×10 mm×1.1 mm)作为工作电极,0.1 mol/L NaOAc/NaOH作电解质溶液,将扫描速度固定为100 mV/s,分别记录不同pH值(9.0~13.0)和不同配合物浓度(0.2~1.0 mmol/L)的循环伏安(CV)曲线。
1.3.2 控制电压电解控制电压电解采用三电极系统:ITO导电玻璃作为工作电极;Ag/AgCl作参比电极;Pt片(10 mm×10 mm)作对电极。电解液为pH=12.4的0.1 mol/L NaOAc/NaOH溶液。通过施加不同的催化电压,记录5 min时的电流读数,并绘制电流密度与过电位的关系曲线(Tafel曲线)。
1.4 产氧测试和法拉第效率的计算电化学池选用全密封双通道H型光电化学池,阳极室和阴极室通过玻璃砂芯分隔。阳极室装有工作电极、参比电极(Ag/AgCl)和实时氧气监测探针。阴极室装有铂片作对电极。取pH=12.4的0.1 mol/L NaOAc/NaOH溶液作为电解液分别加入阴极室和阳极室,并向阳极室中加入[Cu(THEED)(H2O)]SO4配成1.0 mmol/L的溶液。通N2气20 min排除电化学池中的氧气后,将电化学池密封。施加1.35 V(相对于标准氢电极)电压,同时使用氧气监测探针对电化学池顶部的氧气含量进行检测。通过法拉第定律计算出理论产氧量和法拉第效率。
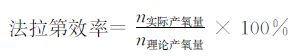
使用扫描电子显微镜(SEM,Hitachi-4800场发射扫描电子显微镜,电压5.0 kV)对所形成的活性CuO膜进行表面形貌表征,并用能量色散X射线(EDX)进行元素分析。X射线衍射(XRD)采用Bruker D8 Focus X射线衍射仪,用于活性膜的晶体结构分析。多功能光电子能谱仪(XPS,PHI Quantera SXM)确定Cu元素的价态及存在形式。
2 结果与讨论 2.1 铜配合物的晶体结构甲醇和水(1∶1)作为溶剂,采用乙醚扩散法培养铜配合物的晶体。通过X射线单晶衍射分析测定其结构(CCDC号:1049629),晶体结构参数列于表 1中。X射线研究表明配合物呈八面体构型。以Cu为中心,THEED上的2个N原子、3个O原子和1个水分子中的O原子与Cu配位(图 1)。
|
|
表1 [Cu(THEED)(H2O)]SO4晶体参数 X-ray crystallographic parameters of [Cu(THEED)(H2O)]SO4 |

|
图1 [Cu(THEED)(H2O)]2+的晶体结构图 Crystal structure of [Cu(THEED)(H2O)]2+ |
用0.1 mol/L NaOAc/NaOH作溶液配制1.0 mmol/L [Cu(THEED)(H2O)]SO4。用浓NaOH调节其pH值到9.0~13.0之间,并记录相应的循环伏安曲线。如图 2a所示,随pH值的逐渐增加,催化电流不断增大。固定pH=12.4,从0.2 V向1.5 V扫描,循环伏安曲线出现一个较大的不可逆氧化峰。该峰起始于1.0 V处,是水的氧化峰。其催化电流随催化剂含量的增加而呈上升趋势(图 2b)。在1.3 V处电流大小与催化剂浓度符合线性关系(图 2c)。扩大扫描范围在-1.0~1.5 V之间,使用ITO导电玻璃作为工作电极连续多次进行循环伏安扫描(图 2d)。阳极产生的氧化电流随循环次数的增加而上涨,这一现象暗示有活性物质在阳极表面富集。阴极-0.3 V处也出现一个随循环次数增加而不断变大的峰,根据文献该峰是氧气的还原峰(O2/O-2)。此峰的出现及其电流密度的上升是因为不断有氧气生成[17],这进一步证明水被氧化。

|
图2 [Cu(THEED)(H2O)]SO4在不同条件下的循环伏安图0.10 mol/L NaOAc/NaOH,玻璃碳电极(S= 0.07 cm2) (a)1.0 mmol/L [Cu(THEED)(H2O)]SO4,不同pH值的循环伏安曲线;(b)pH = 12.4,不同浓度[Cu(THEED)(H2O)]SO4的循环伏安曲线;(c)催化剂浓度与1.3 V处电流的关系;(d)1.0 cm2 ITO玻璃电极,1.0 mmol/L [Cu(THEED)(H2O)]SO4,pH = 12.4,多次循环伏安曲线 CVs of [Cu(THEED)(H2O)]SO4 in different conditions 0.10 mol/L NaOAc/NaOH,on a glassy carbon electrode (S=0.07 cm2) (a)CVs of 1.0 mmol/L of [Cu(THEED)(H2O)]SO4 at various pH,(b) [Cu(THEED)(H2O)]SO4 at various concentrations at pH=12.4,(c) the relationship between the catalyst concentrations and catalytic currents at 1.3 V, (d)multiple CVs at a 1.0 cm2 ITO glass,1.0 mmol/L [Cu(THEED)(H2O)]SO4 at pH=12.4 |
控制电压为1.35 V,电解溶有1.0 mmol/L[Cu(THEED)(H2O)]SO4的NaOAc/NaOH溶液(0.1 mol/L,pH=12.4)。电解过程中可以观察到在ITO电极表面不断有气泡生成,并有黑色固体在其表面富集。随着时间的延长,ITO电极表面的黑色物质逐渐增加,相应的电流密度也从0.3 mA/cm2增加到1.4 mA/cm2(图 3a)。据此我们推断黑色物质的累积有助于电催化反应的进行。经过5 h的电解共产生氧气30 μmol,法拉第效率为81 %(图 3b)。

|
图3 控制电压电解[Cu(THEED)(H2O)]SO4并产氧 控制电压1.35 V,电解1.0 mmol/L [Cu(THEED)(H2O)]SO4,pH = 12.4,0.10 mol/L NaOAc/NaOH溶液 (a)电流密度时间曲线;(b)时间响应产氧曲线 Controlled potential electrolysis of [Cu(THEED)(H2O)]SO4 and oxygen evolution Controlled potential electrolysis at 1.35 V,1.0 mmol/L [Cu(THEED)(H2O)]SO4 in 0.10 mol/L NaOAc/NaOH at pH=12.4 (a) Time dependence of current density curve,(b) time dependence of O2 amount |
用去离子水小心冲洗附着在ITO玻璃上的黑色薄膜,然后将其置于真空干燥箱中干燥过夜。SEM显示活性CuO膜的表面形貌类似于Co-Pi产氧催化剂[22]:球状小颗粒相互堆积成簇附着于ITO电极表面,颗粒大小约为2 μm(图 4a)。EDX结果显示这些黑色颗粒的组成元素以Cu和O为主(图 4b)。未检测到C和N则表明配体和电解质均未对活性物质的组成产生影响。因此,以[Cu(THEED)(H2O)]SO4作为前驱体产生的活性物质既不同于6,6′-二羟基-2,2′-联吡啶铜电沉积产生的多聚或寡聚物[23],也不同于铜盐在硼酸缓冲溶液中产生的Cu-Bi膜[21]。事实上,它更类似于用铜二甲氨基吡啶配合物作为前驱体制备的活性CuO[20]。图 4c为活性膜的XRD图。我们只检测到了ITO的特征衍射峰但没有观察到CuO的衍射峰,所以活性物质应该是无定形的。在铜的高分辨XPS图中(图 4d),933.6和953.6 eV分别对应Cu 2p3/2和Cu 2p1/2的结合能。由此判定Cu的化合价为+2价,主要组成是CuO,并伴随有Cu(OH)2。两个主峰之间的卫星峰可进一步证明CuO的存在[20, 24, 25]。

|
图4 活性CuO的表征 (a)SEM图;(b)EDX图;(c)XRD 图;(d)Cu 2p的XPS图 Characterization of active CuO (a) SEM image,(b) EDX image,(c) XRD image,(d) XPS image of Cu 2p |
为研究CuO膜的催化活性,我们进行了循环伏安测试和控制电压电解实验。实验均在不含Cu(Ⅱ)、pH=12.4、0.1 mol/L的NaOAc/NaOH溶液中进行。
循环伏安测试如图 5a所示,不同于[Cu(THEED)(H2O)]SO4溶液(图 2d),0.78 V的位置上出现了一个新的不可逆还原峰;-0.13 V处出现了新的氧化峰。根据文献可将-0.13 V处的氧化峰归属于Cu(Ⅱ/Ⅰ)[23]。在该条件下,活性CuO的过电位约为500 mV。经过50次循环,电流值下降了10%。主要原因在于循环过程中CuO出现部分剥落的情况。同时,由于电解过程中并未搅拌,所以电极附近pH值的降低也是电流减小的原因之一。

|
图5 CuO膜的活性 0.10 mol/L NaOAc/NaOH,pH = 12.4 (a)附着有CuO的ITO电极第1次和第50次循环的伏安曲线;(b)过电位与电流密度关系曲线(Tafel 曲线), 内图为附着有CuO的ITO电极照片;1.35 V电压(c)电解附着有CuO的ITO和空白ITO电极;(d)时间响应产氧曲线 Activity of CuO film 0.10 mol/L NaOAc/NaOH at pH=12.4 (a) CV curves recorded of the 1st cycle and the 50th cycle for CuO covered on ITO glass,(b) the overpotential varied with the current density (Tafel curve),inset: the picture of ITO with CuO covered on; maintained apply potential at 1.35 V, (c) bulk electrolysis ITO covered with CuO and blank ITO,(d) time dependence of O2 amount |
图 5b是活性CuO膜的Tafel曲线:当过电位在0.3~0.6 V之间时,电流密度指数随过电位线性变化,Tafel斜率为147 mV/decade。当过电位超过0.6 V后,增加电压,电流不再明显增大。设置催化电压为1.35 V,催化电流密度可维持在1.5 mA/cm2 近6 h不变(图 5c),其稳定性要远远高于Cu盐在碳酸钠溶液中形成的不稳定薄膜(15 min溶解于碳酸溶液中)[19]。电解7.5 h共产生氧气97 μmol,法拉第效率为95 %(图 5d)。
3 结论本文以廉价易得的无水CuSO4和商业化试剂THEED为原料,通过简单的方法合成了在水中具有良好溶解性的[Cu(THEED)(H2O)]SO4配合物。电解溶有[Cu(THEED)(H2O)]SO4的碱性溶液,可以在阳极上原位制备具有产氧催化活性的CuO膜。研究表明使用[Cu(THEED)(H2O)]SO4作为催化剂前驱体可以有效的避免碱性条件下Cu(OH)2沉淀的产成。使用上述方法原位生成的CuO可以在碱性条件下高效地电催化水氧化。在pH=12.4的溶液中,其过电位约500 mV,法拉第效率高达95%。与此同时,该催化剂具有良好的稳定性。经过长时间的电解,催化电流密度能保持不变。
| [1] | Lewis N S,Nocera D G. Powering the planet: chemical challenges in solar energy utilization[J]. Proceedings of the National Academy of Sciences of the United States of America,2006, 103: 15729-15735. |
| [2] | 李秋叶,金振声. 可见光"全"分解水的类纳光电化学(PEC)电池模型[J]. 影像科学与光化学,2015, 33(2): 99-107. Li Q Y, Jin Z S. Nano photoelectrochemical cell-like model for visible-light-responded overall splitting of water[J]. Imaging Science and Photochemistry, 2015, 33(2): 99-107. |
| [3] | Eisenberg R, Gray H B. Preface on making oxygen[J]. Inorganic Chemistry, 2008, 47: 1697-1699. |
| [4] | Kärkäs M D, Verho O, Johnston E V, kermark B. Artificial photosynthesis: molecular systems for catalytic water oxidation[J]. Chemical Reviews, 2014, 114(24): 11863-12001. |
| [5] | Gersten S W,Samuels G J, Meyer T J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer[J]. Journal of the American Chemical Society, 1982, 104: 4029-4030. |
| [6] | Lee Y, Suntivich J, May K J, Perry E E, Shao-Horn Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions[J]. Journal of Physical Chemistry Letters, 2012, 3: 399-404. |
| [7] | Blakemore J D, Mara M W, Kushner-Lenho M N, Schley N D, Konezny S J, Rivalta I, Negre C F A, Snoeberger R C, Kokhan O, Huang J, Stickrath A, Tran L A, Parr M L, Chen L X,Tiede D M, Batista V S, Crabtree R H, Brudvig G W. Characterization of an amorphous iridium water-oxidation catalyst electrodeposited from organometallic precursors[J]. Inorganic Chemistry, 2013, 52: 1860-1871. |
| [8] | Dismukes G C, Brimblecombe R, Felton G A N, Pryadun R S, Sheats J E, Spiccia L, Swiegers G F. Development of bioinspired Mn4O4-cubane water oxidation catalysts: lessons from photosynthesis[J]. Accounts of Chemical Research, 2009, 42: 1935-1943. |
| [9] | Kanan M W, Nocera D G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+[J]. Science, 2008, 321:1072-1075. |
| [10] | Han A L, Wu H T, Sun Z J, Jia H X, Du P W. Facile deposition of nanostructured cobalt oxide catalysts from molecular cobaloximes for efficient water oxidation[J]. Physical Chemistry Chemical Physics, 2013, 15:12534-12538. |
| [11] | Han A L, Wu H T, Sun Z J, Jia H X, Yan Z P, Ma H, Liu X, Du P W. Green cobalt oxide (CoOx) film with nanoribbon structures electrodeposited from the BF-2 annulated cobaloxime precursor for efficient water oxidation[J]. ACS Applied Materials & Interfaces, 2014,6:10929-10934. |
| [12] | Dinc M, Surendranath Y, Nocera D G. Nickel-borate oxygen-evolving catalyst that functions under benign conditions[J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107: 10337-10341. |
| [13] | Singh A, Chang S L Y, Hocking R K, Bach U, Spiccia L. Highly active nickel oxide water oxidation catalysts deposited from molecular complexes[J]. Energy & Environment Science, 2013, 6: 579-586. |
| [14] | Wang D,Ghirlanda G, Allen J P. Water oxidation by a nickel-glycine catalyst[J]. Journal of the American Chemical Society, 2014, 136: 10198-10201. |
| [15] | Fillol J L,Codolà Z, Garcia-Bosch I, Gómez L, Pla J J, Costas M. Efficient water oxidation catalysts based on readily available iron coordination complexes[J]. Nature Chemistry, 2011, 3: 807-813. |
| [16] | Li T T, Cao S, Yang C, Chen Y, Lv X J, Fu W F. Electrochemical water oxidation by in situ-generated copper oxide film from [Cu(TEOA)(H2O)2][SO4] complex[J]. Inorganic Chemistry, 2015, 54: 3061-3067. |
| [17] | Barnett S M, Goldberg K I, Mayer J M. A soluble copper-bipyridine water-oxidation electrocatalyst[J]. Nature Chemistry, 2012, 4: 498-502. |
| [18] | Zhang M T, Chen Z F, Kang P, Meyer T J. Electrocatalytic water oxidation with a copper(Ⅱ) polypeptide complex[J]. Journal of the American Chemical Society, 2013, 135: 2048-2051. |
| [19] | Chen Z F, Meyer T J. Copper(Ⅱ) catalysis of water oxidation[J]. Angewandte Chemie International Edition, 2013, 52: 700-703. |
| [20] | Liu X, Jia H X, Sun Z J, Chen H Y, Xu P, Du P W. Nanostructured copper oxide electrodeposited from copper(Ⅱ) complexes as an active catalyst for electrocatalytic oxygen evolution reaction[J]. Electrochemistry Communication, 2014, 46: 1-4. |
| [21] | Yu F S, Li F, Zhang B B, Li H, Sun L C. Efficient electrocatalytic water oxidation by a copper oxide thin film in borate buffer[J]. ACS Catalysis, 2015, 5: 627-630. |
| [22] | Surendranath Y, Dincǎ M, Nocera D G. Electrolyte-dependent electrosynthesis and activity of cobalt-based water oxidation catalysts[J]. Journal of the American Chemical Society, 2009, 131: 2615-2620. |
| [23] | Zhang T, Wang C, Liu S B, Wang J L, Lin W B. A biomimetic copper water oxidation catalyst with low overpotential[J]. Journal of the American Chemical Society, 2014, 136: 273-281. |
| [24] | Du J L, Chen Z F, Ye S R, Wiley B J, Meyer T J. Copper as a robust and transparent electrocatalyst for water oxidation[J]. AngewandteChemie International Edition, 2015, 54: 2073-2078. |
| [25] | Casella I G, Gatta M. Anodic electrodeposition of copper oxide/hydroxide films by alkaline solutions containing cuprous cyanide ions[J]. Journal of Electroanalytical Chemistry, 2014, 494(2000): 12-20. |