




2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, P. R. China
亚砜类化合物具有广谱生物活性[1],经结构修饰以后,可广泛应用于农药和医药领域,选择性的氧化硫醚是制备亚砜的常用方法之一。硫醚过氧化会产生砜,因此,如何控制氧化反应停留在亚砜阶段极其重要。为了得到高选择性的硫醚氧化产物,多种氧化剂和大量的氧化方法被尝试,从传统的使用高锰酸钾、二氧化硒和硝酸等强氧化剂氧化,到有机分子[2]或过渡金属[3-6]催化的双氧水氧化等,均取得了一定的研究成果,但是依然存在选择性差、废弃物对环境污染严重、氧化剂使用量不易控制等问题。
光催化氧化反应是实现绿色化学可持续发展的途径之一。近几年,无机半导体光催化氧化反应在硫醚的选择性氧化中取得了瞩目的研究成果,但是仍然存在下列问题:光照过程中同时出现的·OH、·OOH和空穴等活性成分[7],有可能造成亚砜的继续氧化或其它基团的氧化;部分体系中用到贵金属Pt[8];TiO2体系需要外加三乙胺或染料来提高催化活性和选择性[9, 10],不利于产物的提纯和分离;单独的mpg-C3N4[11]催化效率和选择性都不理想,虽然使其与CdS复合后得到的CdS/C3N4[12]效率有所提高,但是CdS废弃后会污染环境。研究异相体系的同时,均相光催化氧化硫醚体系也在快速发展。目前,基于有机染料和金属配合物的光催化氧化硫醚在催化机理和转化率上有所突破,但是也面临贵金属配合物(Pt[13]、Ru[14-19])催化体系因价格昂贵与实际应用不相称、染料体系的废弃物对环境有害等问题。因此,均相光催化氧化硫醚体系仍然需要引入高效、廉价、清洁的光敏剂,适宜的光敏剂可以把氧化能力较弱的氧气分子(O2)通过电子转移或激发态能量转移来形成相应的氧化能力较强的超氧自由基和单重态氧(1O2),利用这两个中间体可以实现硫醚到亚砜的高选择性氧化[20]。
苝和苝酐被广泛用于有机合成和染料的制备,而对它们用在均相光催化反应体系中的报道却很少[21]。本文将苝酐进行化学修饰后得到PMI-(iPr)2An(PSⅡ)和PMI-((iPr)2An)2(PSⅢ),结构式如图 1所示。二者相比于苝(Perylene,PSⅠ),对可见光的吸收范围红移;相对于苝酐,溶解性变好。为了拓展苝衍生物的应用范围,本文将PSⅡ和PSⅢ用于光催化氧化硫醚,经过对活性中间体的清除实验和电子顺磁共振谱(ESR)等,显示该体系中同时存在单重态氧(1O2)和超氧自由基;在优化条件下,光敏剂用量为0.1%(摩尔分数)时,PSⅢ表现出高的催化氧化效率、优异的选择性和对苯基硫醚衍生物的普适性。与报道的贵金属配合物相比,光敏剂价格低廉;与曙红[22]、罗丹明B[22]和维生素B2衍生物[23](摩尔分数2%)相比,本体系中光敏剂用量大大缩减且不含重原子(Br、I),无需额外加HCl,利于催化剂成本的降低和产物的提纯。

|
图 1 光敏剂PSⅠ、PSⅡ和PSⅢ的化学结构 Fig.1 Chemical structures of PSⅠ, PSⅡ and PSⅢ |
仪器:BrukerBiflexⅢ型质谱仪;LCMS-IT/TOF高效液相-电喷雾-离子阱/飞行时间串联质谱仪(ShimadzuJapan);400 MHz Bruker核磁共振仪;Hitachi UV-3010紫外可见吸收光谱仪;Hitachi F-4500荧光光谱仪;CHI660C电化学工作站(上海辰华仪器公司);Bruker ESP-300E电子顺磁共振仪。
试剂:4-甲氧基茴香硫醚、3-甲氧基茴香硫醚、2-甲氧基茴香硫醚、对溴茴香硫醚、对羟基茴香硫醚、对硝基茴香硫醚、甲基苯基硫醚,百灵威公司;苝、3, 4, 9, 10-苝四甲酸二酐、2, 6-二异丙基苯胺,希恩思产品;CDCl3、重水、重氧水,阿拉丁;2, 2, 6, 6-四甲基哌啶(TEMP)、5, 5-二甲基-1-吡咯啉氮氧化物(DMPO)、四丁基六氟磷酸铵([Bu4N][PF6])、对苯二酚,安耐吉;叔丁醇、醋酸锌、咪唑、盐酸、无水乙醇、乙腈、二氯甲烷、N, N-二甲基甲酰胺(DMF)、硝基甲烷,均购于北京化工厂;所用水均为Milli-Q超纯水(> 18 MΩ)。所有用于光催化反应、光谱、电化学等测试的试剂均直接使用,未经进一步处理。
1.2 光敏剂的合成光敏剂PSⅡ和PSⅢ均参考文献[24]方法合成。
PSⅡ的合成:将19 g 3, 4, 9, 10-苝四甲酸二酐、4.3 g 2, 6-二异丙基苯胺、7g醋酸锌、90 g咪唑和40 mL水,在100 mL的反应釜里混合均匀以后,190 ℃下反应24 h。冷却至室温后,向混合液中加入20 mL浓盐酸,得到固/液混合物。过滤收集固体,用蒸馏水多次洗涤干燥,得到粗产物。色谱柱提纯粗产物后得到5.3 g红色产物(产率22.7%)。1HNMR (CDCl3, 400 MHz): 8.67(d, J=8.0 Hz, 2H), 8.48(t, J=8.0Hz, 4H), 7.93(d, J=8.0 Hz, 2H), 7.66(t, J=8.0 Hz, 2H), 7.48(t, J=8.0 Hz, 1H), 7.34(d, J=8.0 Hz, 2H), 2.77(sep, 2H), 1.18(d, 12H)。
PSⅢ的合成:与PSⅡ类似,只将3, 4, 9, 10-苝四甲酸二酐和2, 6-二异丙基苯胺的用量分别改为2 g和4 g,其他条件保持不变。1HNMR (CDCl3, 400 MHz): 8.78 (dd, 8 H), 7.51(m, 2 H), 7.36 (d, 4 H), 2.76 (m, 4 H), 1.20 (d, 24 H)。
1.3 循环伏安和光谱测试用打磨好的玻碳电极(面积0.07 cm2)作工作电极,Pt片(面积1 cm2)作对电极,Ag/AgCl作参比电极,加适量二茂铁(Fc)作内标,0.1 mol/L [Bu4N][PF6]氩气饱和的DMF溶液作电解质,扫描速度固定为100 mV/s, 分别记录PSⅠ、PSⅡ和PSⅢ(1.0 mmol/L)的循环伏安(CV)曲线。
3个光敏剂分别配成0.01 mmol/L的DMF溶液,测吸收光谱和发射光谱。用Rehm-Weller公式[25]计算吉布斯自由能(式1)。
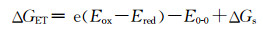
|
(1) |
计算电子从硫醚转移到光敏剂激发态的吉布斯自由能时,式中Eox是硫醚的氧化电位,Ered为光敏剂的还原电位。计算电子从光敏剂激发态转移到氧气的吉布斯自由能时, Eox代表光敏剂的氧化电位,Ered为O2到超氧自由基的还原电位。在极性溶剂中,ΔGs可以忽略不计。
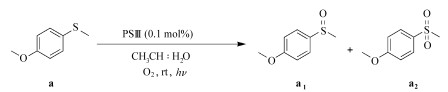
1.4 光催化氧化硫醚本文中的光催化氧化实验主要包括:使用PSⅡ初步筛选反应条件;比较和评估3个光敏剂在同一条件下的催化性能;优化混合溶剂(乙腈:水)的比例;优化PSⅢ和底物的浓度;改变不同的硫醚底物,评估PSⅢ的普适性;机理实验。
基本的实验操作和条件:将硫醚、光敏剂和溶剂加入15 mL的玻璃试管中,利用一个大气压的O2饱和溶液后封管并光照(LEDs, 3W*30, λ≥ 420 nm)一定时间;所有催化反应均在室温下进行,溶剂总体积均为4.5 mL(除特殊说明以外,均是按以上条件进行)。
光照结束后,用旋转蒸发仪除去溶剂得到混合物,借助混合物的核磁共振氢谱来确定硫醚的转化率和亚砜的选择性(硝基甲烷作内标,CDCl3作溶剂)。底物中与硫原子相连的甲基上的氢在核磁共振氢谱中的化学位移为2.40,产物为亚砜时该化学位移为2.72,产物为砜时该化学位移为3.05。转化率和选择性按以下的公式计算:

|
(2) |
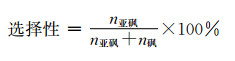
|
(3) |
图 2为PSⅠ、PSⅡ和PSⅢ归一化的吸收光谱和发射光谱。从图中可以看出,在苝酐的一端或两端修饰同种类型的基团以后,得到的衍生物PSⅡ和PSⅢ相对于苝对可见光的吸收范围向长波方向移动。苝吸光范围为360~450 nm,其中部分属于紫外光区;苝酐的一端引入基团得到的PSⅡ,其吸收范围为390~550 nm;苝酐的两端同时引入基团后得到的PSⅢ,其吸收范围为440~550 nm。PSⅡ和PSⅢ在最大吸收波长处对应的摩尔消光系数分别为33900和84200 mol-1·L·cm-1,二者有强的可见光吸收能力。从光敏剂(PSⅠ、PSⅡ和PSⅢ)的吸收光谱和发射光谱的交点分别得出相应的λ0-0。

|
图 2 (a)归一化的PSⅠ的吸收光谱和发射光谱;(b)归一化的PSⅡ的吸收光谱和发射光谱;(c)归一化的PSⅢ的吸收光谱和发射光谱吸收光谱和发射光谱均在DMF中测定(0.01 mmol/L),激发波长365 nm Fig.2 Normalized UV-Vis absorption and emission spectra of (a) PSⅠ, (b) PSⅡ, (c) PSⅢ in DMF (0.01 mmol/L, λex = 365 nm) |
用0.1 mol/L [Bu4N][PF6]的DMF溶液配制1.0 mmol/L光敏剂溶液,分别记录氩气饱和后的相应循环伏安(CV)曲线,3个光敏剂在基态和激发态时的氧化还原电势见表 1。甲基苯基硫醚的氧化电势为0.012 V (vs.Fc+/0)[26],用Rehm-Weller公式计算电子从甲基苯基硫醚转移到光敏剂激发态的吉布斯自由能,对应于PSⅠ、PSⅡ和PSⅢ的吉布斯自由能分别是-0.65 eV、-0.99 eV和-1.13 eV。一些敏化剂经光照激发以后可以把电子转移给O2形成超氧自由基,超氧自由基可以氧化硫醚,所以计算这一途径的吉布斯自由能也是有必要的。O2到超氧自由基的还原电势为-0.91 V (vs. Fc+/0)[11],PSⅠ、PSⅡ和PSⅢ经激发态电子转移产生超氧自由基时,对应的吉布斯自由能分别为-1.34 eV、-0.64 eV和-0.28 eV。两种情况下的吉布斯自由能均小于零,说明电子从硫醚转移到光敏剂激发态、电子从光敏剂激发态转移到O2在热力学上均可行。
| 表 1 光敏剂在基态和激发态的氧化还原电势 Table 1 Redox potentials of PS and PS* |
利用光敏剂PSⅡ来初步筛选光催化氧化硫醚的条件,通过改变溶剂种类、光敏剂用量、有无光照等变量设计10个平行实验(表 2)。6、7、8、9和10的结果说明:PSⅡ在光照和氧气的条件下能把硫醚氧化成亚砜,且光照、氧气、光敏剂三者缺一不可。1~6项结果说明,该体系依赖于溶剂的种类,乙腈作溶剂更利于硫醚的氧化,向乙腈中加入适量的水,可以提高硫醚的转化率。6和7的结果表明,光敏剂的剂量可以小至0.67 %(光敏剂/底物,摩尔分数)。
| 表 2 光催化氧化4-甲氧基茴香硫醚条件的初步优化 Table 2 The preliminary optimization of the experimental conditions forphotooxidation of 4-methoxythioanisole |
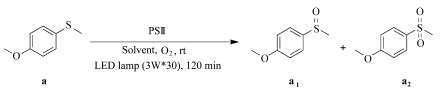
为了比较PSⅠ、PSⅡ和PSⅢ的催化活性,在同样的反应条件下进行光催化氧化硫醚实验,结果如表 3所示。三者中,PSⅢ对硫醚的催化氧化效果最佳,硫醚的转化率为97.8%,而PSⅡ和PSⅠ的转化率仅为39.8%和34.0%。
| 表 3 PSⅠ、PSⅡ和PSⅢ光催化氧化4-甲氧基茴香硫醚能力的比较 Table 3 Comparison of the photocatalytic ability of PSⅠ, PSⅡ and PSⅢ for photooxidationof 4-methoxythioanisole |
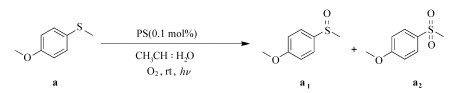
乙腈中加入适量的水可以促进硫醚的氧化(表 2中5、6项),因此探究乙腈和水的用量比对催化效率的影响很重要。固定PSⅢ与底物的摩尔比为0.1%,进行不同体积比CH3CN:H2O(4.5:0、89:1、17:1、8:1、7:2、2:1) 下的光催化反应,结果见表 4。当CH3CN:H2O为7:2,硫醚到亚砜的转化率最高,水的用量比太大或太少都不利于催化氧化,且水太多会导致光敏剂从反应液中析出。
| 表 4 PSⅢ在CH3CN与H2O不同比例下光催化氧化4-甲氧基茴香硫醚的结果 Table 4 Photocatalytic oxidation of 4-methoxythioanisole with PSⅢ as photocatalyst under different proportions of CH3CN and H2O |
光敏剂和底物的浓度会对光催化的效率有影响。固定PSⅢ与底物的摩尔比为0.1%,同时改变硫醚和光敏剂的浓度来评估催化效率,结果如表 5所示。PSⅢ和硫醚的浓度同时增大时,转化率和单位时间内的转化率(TOF)呈下降趋势,说明高浓度的光敏剂和底物不利于硫醚的氧化,而低浓度的光敏剂和硫醚更利于砜的生成。实验中,光敏剂和底物的最低浓度分别至0.0069 mmol/L和6.9 mmol/L。更低的浓度或许会达到更高的转化率,但会使溶剂的使用量增加。
| 表 5 不同浓度下光催化氧化4-甲氧基茴香硫醚的结果 Table 5 Photocatalytic oxidation of 4-methoxythioanisoleatvarious concentrations |

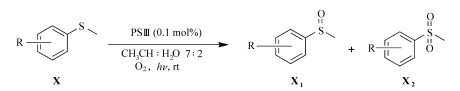
为了研究PSⅢ对其它硫醚底物的适用性,在优化的反应条件下,我们把不同的硫醚衍生物也用在该体系的光催化氧化反应中。刚好完全转化时(对硝基茴香硫醚除外)所用时间如表 6所示。由表 6可知,除了对硝基茴香硫醚没有取得好的转化率,其它所选的硫醚都取得了好的转化率;催化效率受底物上的基团种类和基团位置的影响。PSⅢ对硝基茴香硫醚没有取得好的催化效率,暗示该反应涉及到自由基历程,因为硝基是一个自由基清除剂。
| 表 6 PSⅢ用于硫醚衍生物的光催化氧化效果 Table 6 Photocatalytic oxidation of various sulfides with PSⅢ as photocatalyst |
为进一步研究该体系中光催化氧化硫醚起效果的活性物种和实验机理,我们在反应体系中额外加入不同的化学清除剂(底物的两个当量)来进行活性物种的捕获实验,结果如表 7所示。从表 7可知,第4项中加入2, 2, 6, 6-四甲基哌啶(TEMP)以后,硫醚的氧化受到抑制。TEMP能够清除光照过程中产生的单重态氧(1O2),由此推断该过程中有1O2产生。与不含重原子的普通溶剂相比,重原子取代的溶剂更能稳定1O2,因此要提高以1O2为活性物种的反应效率,可以让反应在含重原子取代的溶剂中进行[27]。为进一步说明1O2参与了PSⅢ为光敏剂的硫醚氧化,我们采用重水(D2O)来代替反应体系中的H2O。第6项和第7项的结果显示,重水可以促进转化率,据此推测该体系中有1O2生成。第6项中加入叔丁醇以后,硫醚到亚砜的转化并没有受到抑制,可以因此认为没有·OH的产生。对苯二酚作为自由基清除剂以及5, 5-二甲基-1-吡咯啉氮氧化物(DMPO)作为超氧自由基的清除剂也被用于该体系。第2项中底物转化率仅为17.3%,间接说明了该反应涉及到自由基反应历程;第5项中底物的转化率也只有41.0%,说明超氧自由基参与了反应。
| 表 7 存在清除剂或助反应剂的机理实验 Table 7 Mechanistic experiments in the presence of scavengers and enhancers |

电子自旋共振(ESR)实验进一步证实该体系中有1O2和超氧自由基。TEMP和DMPO能够分别有效地清除1O2和超氧自由基,是因为这两个清除剂分别与1O2和超氧自由基形成了具有特殊电子顺磁信号的加和物,这两个信号给出了1O2和超氧自由基存在的有力证据。用LED灯(3W*30,λ≥ 420 nm)分别对PSⅢ与DMPO、PSⅢ与TEMP的混合物进行光照。与光照前相比,光照以后观察到了明显的超氧自由基与DMPO加和物的信号(见图 3a)和1O2与TMEP加和物的信号(见图 3c),这充分说明了PSⅢ在有氧光照条件下,会同时产生超氧自由基和1O2,同时也证实了前面用Rehm-Weller公式计算得出的结果——电子从PSⅢ的激发态转移到氧气形成超氧自由基是自发过程。在有硫醚存在的条件下,相应的超氧自由基(见图 3b)和1O2(见图 3d)信号都减弱了,说明底物会消耗超氧自由基和1O2,即超氧自由基和1O2都在硫醚氧化过程中发挥作用,这与表 7中的清除活性物种的实验结果相符合。

|
图 3 不同混合物的电子自旋共振谱(a) PSⅢ (0.01 mmol/L)和DMPO (10 mmol/L),光照4 min;(b)PSⅢ (0.01 mmol/L)、DMPO (10 mmol/L)和4-甲氧基茴香硫醚(3 mmol/L),光照4 min;(c)PSⅢ (0.1 mmol/L)和TEMP(100 mmol/L),光照3 min;(d)PSⅢ (0.1 mmol/L)、TEMP (100 mmol/L)和4-甲氧基茴香硫醚(3 mmol/L),光照3 min所用溶剂均是氧气饱和的CH3CN:H2O (7:2,V/V), 室温,光源(LEDs,3W*30,λ≥420 nm) Fig.3 ESR spectra under different experimental conditions (a) PSⅢ (0.01 mmol/L), DMPO (10 mmol/L), irradiation for 4 min; (b)PSⅢ (0.01 mmol/L), DMPO (10 mmol/L), 4-methoxythioanisole(3 mmol/L), irradiation for 4 min; (c) PSⅢ (0.1 mmol/L), TEMP(100 mmol/L), irradiation for 3 min; (d) PSⅢ (0.1 mmol/L), TEMP(100 mmol/L), 4-methoxythioanisole(3 mmol/L), irradiation for 3 min The solvent (CH3CN:H2O=7:2, V/V) was saturated with O2, LED lamp (3W*30), at room temperature |
适量的H2O可以促进转化率,所以产物亚砜中的O可能来源于H2O。为了探究O的来源,以4-甲氧基茴香硫醚为底物,使用H2O和重氧水H2O18分别进行O的同位素标记。待底物完全转化以后,利用高分辨质谱(ESI IT-TOF)定性检测产物。结果如图 4所示,并未检测到含O18的亚砜产物,因此推测亚砜中的O来自O2。

|
图 4 使用H2O18和H2O时相应产物的高分辨质谱图 Fig.4 Mass spectra of the corresponding sulfoxide obtained by using H2O18 and H2O, respectively |
以上机理实验表明,该体系中硫醚的氧化可能存在两条路径:(1) PSⅢ吸收光以后变为PSⅢ*,PSⅢ*把能量转移给O2形成1O2,光敏剂激发态猝灭,1O2与R2S形成R2S+-OO-,R2S+-OO-与另一分子的R2S反应,生成亚砜;(2) PSⅢ吸收光以后变为PSⅢ*,PSⅢ*把电子转移给O2形成超氧自由基和PSⅢ*+,R2S把电子给PSⅢ*+使PSⅢ*+猝灭,失去电子的R2S被超氧自由基氧化成亚砜。
3 结论本文经过对苝酐进行化学修饰,合成了两个不含金属和重原子的廉价苝衍生物PMI-(iPr)2An(PSⅡ)和PMI-((iPr)2An)2(PSⅢ),其较好的溶解性和强的可见吸收能力利于苝衍生物应用范围的拓展。在可见光照射下,以PSⅡ或PSⅢ作为光敏剂,氧气为氧化剂可以实现高转化率和高选择性的硫醚到亚砜的氧化。在优化反应条件下,PSⅢ对苯基硫醚衍生物光催化氧化的转化率和选择性均接近100%。活性中间体的清除实验和电子顺磁共振实验表明该体系中单重态氧(1O2)和超氧自由基同时对硫醚的氧化起作用。该体系与报道过的贵金属配合物体系相比,价格低廉且效率更高;与曙红、罗丹明B和维生素B2衍生物(摩尔分数2%)相比,其用量小且无需额外加HCl,利于催化剂成本的降低和产物的提纯。该研究可促进以PSⅢ为主的苝衍生物在光催化有机合成中的应用。
| [1] | Legros J, DehliJ R, BolmC. Applications of catalytic asymmetric sulfide oxidations to the syntheses of biologically active sulfoxides[J]. Advanced Synthesis & Catalysis, 2005, 347(1): 19–31. |
| [2] | Huang Y B, Yi W B, Cai C. A recyclable fluorousthioureaorganocatalyst for the chemoselective oxidation of sulfides[J]. Journal of Fluorine Chemistry, 2011, 132(8): 554–557. DOI:10.1016/j.jfluchem.2011.05.026 |
| [3] | Rezaeifard A, Haddad R, Jafarpour M, Hakimi M. {Mo132} nanoball as an efficient and cost-effective catalyst for sustainable oxidation of sulfides and olefins with hydrogen peroxide[J]. ACS Sustainable Chemistry & Engineering, 2014, 2(4): 942–950. |
| [4] | Jabbari A, Mahdavi H, Nikoorazm M, Ghorbani-Choghamarani Arash. Oxovanadium(Ⅳ) salicylideneschiff base complex anchored on mesoporous silica MCM-41 as hybrid materials:a robust catalyst for the oxidation of sulfides[J]. Journal of Porous Materials, 2015, 22(4): 1111–1118. DOI:10.1007/s10934-015-9986-9 |
| [5] | Zhang Z H, Yang X S, Zhang Q Q, Wang L, He M Y, Chen Q, Huang X F. Structure-induced catalysis enhancement of Cu-amino catalysts for rapidly selective oxidation of sulfides in the presence of H2O2[J]. RSC Advances, 2016, 6(106): 104036–104040. DOI:10.1039/C6RA22393A |
| [6] | Wang F, Liu C, Liu G, Li W X, Liu J H. Selective oxidation of sulfides to sulfoxides using hydrogen peroxide over Au/CTN-silica catalyst[J]. Catalysis Communications, 2015, 72: 142–146. DOI:10.1016/j.catcom.2015.09.015 |
| [7] | Kisch H. Semiconductor photocatalysis-mechanistic and synthetic aspects[J]. AngewandteChemie International Edition, 2013, 52(3): 812–847. DOI:10.1002/anie.201201200 |
| [8] | Zhang B, Li J, Zhang B Q, Chong R F, Li R G, Yuan B, Lu S M, Li C. Selective oxidation of sulfides on Pt/BiVO4 photocatalyst under visible light irradiation using water as the oxygen source and dioxygen as the electron acceptor[J]. Journal of Catalysis, 2015, 323: 95–100. |
| [9] | Lang X J, Hao W, Leow W R, Li S Z, Zhao J C, Chen X D. Tertiary amine mediated aerobic oxidation of sulfides into sulfoxides by visible-light photoredox catalysis on TiO2[J]. Chemical Science, 2015, 6(8): 5000–5005. DOI:10.1039/C5SC01813G |
| [10] | Lang X J, Zhao J C, Chen X D. Visible-light-induced photoredox catalysis of dye-sensitized titanium dioxide:selective aerobic oxidation of organic sulfides[J]. Angewandte Chemie International Edition, 2016, 55(15): 4697–4700. DOI:10.1002/anie.201600405 |
| [11] | Zhang P F, Wang Y, Li H R, Antonietti M. Metal-free oxidation of sulfides by carbon nitride with visible light illumination at room temperature[J]. Green Chemistry, 2012, 14(7): 1904–1908. DOI:10.1039/c2gc35148j |
| [12] | Xu Y, Fu Z C, Cao S, Chen Y, Fu W F. Highly selective oxidation of sulfides on a CdS/C3N4 catalyst with dioxygen under visible-light irradiation[J]. Catalysis Science & Technology, 2017, 7(3): 587–595. |
| [13] | Antonio Casado-Sánchez, RocíoGómez-Ballesteros, Tato F, Soriano F J, Gustavo Pascual-Coca, Cabrera S, AlemánJ. Pt(Ⅱ) coordination complexes as visible light photocatalysts for the oxidation of sulfides using batch and flow processed[J]. Chemical Communications, 2016, 52(58): 9137–9140. DOI:10.1039/C6CC02452A |
| [14] | Li T T, Li F M, Zhao W L, Tian Y H, Chen Y, Cai R, Fu W F. Highly efficient and selective photocatalytic oxidation of sulfide by a chromophore-catalyst dyad of ruthenium-based complexes[J]. Inorganic Chemistry, 2015, 54(1): 183–191. DOI:10.1021/ic5020972 |
| [15] | Tanaka H, Nishikawa H, Uchida T, Katsuki T. Photopromoted Ru-catalyzed asymmetric aerobic sulfide oxidation and epoxidation using water as a proton transfer mediator[J]. Journal of the American Chemical Society, 2010, 132(34): 12034–12041. DOI:10.1021/ja104184r |
| [16] | Murugan K S, Rajendarn T, Balakrishnan G, Ganesan M, Sivasubramanian V K, Sankar J, Ilangovan A, Ramamurthy P, Rajagopal S. Visible-light activation of the bimetallic chromophore-catalyst dyad:analysis of transient intermediates and reactivity toward organic sulfides[J]. The Journal of Physical Chemistry A, 2014, 118(25): 4451–4463. DOI:10.1021/jp501084b |
| [17] | Hikita T, Tamaru K, Yamagishi A, Iwamoto T. Production of an optically active sulfoxide by use of colloidally dispersedΛ-tris(2, 2'-bipyridyl)ruthenium(Ⅱ) montmorilloniteas a photosensitizer[J]. Inorganic Chemistry, 1989, 28(11): 2221–2223. DOI:10.1021/ic00310a043 |
| [18] | Zen J M, Liou S L, Kumar A S, Hsia M S. An efficient and selective photocatalytic system for the oxidation of sulfides to sulfoxides[J]. Angewandte Chemie International Edition, 2003, 42(5): 577–579. DOI:10.1002/anie.200390166 |
| [19] | Fujita S, Sato H, Kakegawa N, Yamagishi A. Enantioselective photooxidation of a sulfide by a chiral Ruthenium(Ⅱ) complex immobilized on amontmorillonite clay surface:the role of weak interactions in asymmetric induction[J]. The Journal of Physical Chemistry B, 2006, 110(6): 2533–2540. DOI:10.1021/jp055254r |
| [20] | Bonesi S M, Manet I, Freccero M, Fagnoni M, Albini A. Photosensitized oxidation of sulfides:discriminating between the single-oxygen mechanism and electron transfer involving superoxide anion or molecular oxygen[J]. Chemistry-A European Journal, 2006, 12(18): 4844–4857. DOI:10.1002/(ISSN)1521-3765 |
| [21] | Okamoto S, Kojiyama K, Tsujioka H, Sudo A. Metal-free reductive coupling of C=O and C=N bonds driven by visible light:use of perylene as a simple photoredoxcatalyst[J]. Catalysis Communications, 2016, 52(76): 11339–11342. |
| [22] | Gu X Y, Li X, Chai Y H, Yang Q, Li P X, Yao Y M. A simple metal-free catalytic sulfoxidationundervisible light and air[J]. Green Chemistry, 2013, 15(2): 357–361. DOI:10.1039/c2gc36683e |
| [23] | NeveselýT, SvobodováE, Chudoba J, Sikorski M, Cibulka R. Efficient metal-free aerobic photoxidation of sulfides to sulfoxides mediated by a Vitamin B2 derivative and visible light[J]. Advanced Synthesis & Catalysis, 2016, 358(10): 1654–1663. |
| [24] | Fujitsuka M, Kim S S, Lu C, Tojo S, Majima T. Intermolecular and intramolecular electron transfer processes from excited naphthalene diimide radical anions[J]. The Journal of Physical Chemistry B, 2015, 119(24): 7275–7282. DOI:10.1021/jp510850z |
| [25] | Rehm D, Weller A. Kinetics of fluorescence quenching by electron and H-atom transfer[J]. Israel Journal of Chemi-stry, 1970, 8(2): 259–271. DOI:10.1002/ijch.v8.2 |
| [26] |
黄灵, 赵建章. 具有强可见光吸收的富勒烯-Bodipy衍生物作为光催化剂在硫醚光催化氧化中的应用[J]. 影像科学与光化学, 2014, 32(5): 471–483.
Huang L, Zhao J Z. Fullerene-bodipy dyads and triad showing strong visible light absorption as photocatalysts in photocatalytic oxidation of sulfides[J]. Imaging Science and Photochemistry, 2014, 32(5): 471–483. DOI:10.7517/j.issn.1674-0475.2014.05.471 |
| [27] | Jitka Dad'ová, Svobodová E, Sikorski M, König B, Cibulka R. Photooxidation of Sulfides to Sulfoxides mediated by tetra-o-acetylriboflavin and visible light[J]. ChemCatChem, 2012, 4(5): 620–623. DOI:10.1002/cctc.v4.5 |