




人工模拟光合作用(artificial photosynthesis)是近年来光催化研究领域的热点,其中利用可见光催化还原二氧化碳(CO2),将CO2分子转化为燃料、燃料前体及精细化学品等高附加值产品是人工模拟光合作用研究的重要内容[1-3]。实际上,自然界光合作用系统正是以二氧化碳和水为原料进行有效的“光化学合成”,从而平衡大气层二氧化碳浓度并提供给人类赖以生存的食物及能源。通过可见光催化的方式对二氧化碳进行资源化利用,不仅可以降低二氧化碳的排放量,同时可以满足人类社会日益增长的能源与物质需求。因此,基于对自然界光合作用系统工作原理的认知,构筑人工光合成系统,实现太阳能光催化还原二氧化碳被认为是一种清洁、低能耗、可持续的二氧化碳资源化利用途径[4, 5]。
可见光催化二氧化碳还原体系一般由3个基本组分构建(图 1):用于吸收可见光的光敏剂(photosensitizer)、用于催化还原二氧化碳分子的催化剂(catalyst)、以及用于提供还原反应所需电子的电子牺牲体(sacrificial electron donor)。光敏剂是光催化体系中的“先锋分子”,相当于光合作用中的“天线”分子,它首先吸收可见光产生激发态电子,从而“触发”体系的光催化循环。催化剂是体系中的“灵魂分子”,它是进行二氧化碳催化还原的中心;通常情况下,激发态的光敏剂分子与催化剂分子之间发生光诱导电子转移,生成的还原态催化剂分子才具有催化活性。在理想的人工模拟光合作用体系中,还原二氧化碳所需的电子应该由水的氧化反应来提供,而在目前的研究中,直接将水的氧化与二氧化碳还原在同一体系中耦合仍有困难,因此大多数体系均采用外加易氧化的小分子(胺类、醇类化合物)来作为电子牺牲体。

|
图 1 光催化二氧化碳还原体系示意图 Fig.1 The artificial photosynthetic system for CO2 reduction |
CO2 + 1e-→CO2·- -1.90 V
CO2 +2H+ + 2e-→HCOOH -0.61 V
CO2 +2H+ + 2e-→CO + H2O -0.53 V
2CO2 +2H+ + 2e-→H2C2O4 -0.49 V
CO2 +4H+ + 4e-→HCHO + H2O-0.48 V
CO2 +6H+ + 6e-→CH3OH + H2O-0.38 V
CO2 +8H+ + 8e-→CH4 + 2H2O -0.24 V
二氧化碳是一种化学惰性分子,直接将CO2还原为CO2·-的电势负达-1.9 V (vs. NHE);但是在质子及多电子耦合的条件下,还原CO2的电势可被有效降低。例如,在双电子还原过程中,CO2分子与两个质子耦合被还原为CO和H2O的电势可降至-0.53 V(vs. NHE);当与8个质子耦合被8个电子还原时,CO2可被还原为CH4和H2O,其还原电势进一步降至-0.24 V(vs. NHE)。从以上分析可以看出,二氧化碳还原是一个多电子多质子参与的过程,根据还原程度的高低,还原过程可有2~8个电子参与;同时,由于二氧化碳还原产物多样以及体系不可避免地生成氢气,因此对于二氧化碳还原体系而言还存在产物选择性的问题。对于分子光催化二氧化碳还原体系,体系的转化数(turnover number, 简称TON值)和体系的选择性(selectivity)是评价体系的两个重要参数。
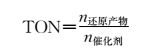
|
(1) |
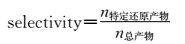
|
(2) |
过渡金属配合物由于具有结构可调控、价态丰富、易于合成等优势,作为一类主要的CO2还原催化剂已长期受到关注。早在20世纪70年代,化学家们已开始以过渡金属配合物作为催化剂来构筑光催化CO2还原体系,在此基础上以过渡金属配合物作为催化剂结合分子光敏剂构筑的均相光催化CO2还原研究得以发展。均相体系具有构筑简单、分子基础明确、便于研究机理的优点,同时应该指出,均相光催化CO2还原体系普遍存在稳定性差、对CO2分子的还原程度低等问题,大多数已报道的均相分子体系在可见光照射下易分解,且仅能将CO2还原为CO或甲酸。
近四十年来,基于Re(Ⅰ)、Ru(Ⅱ)、Ir(Ⅲ)、Mn(Ⅱ)、Cu(Ⅱ)、Fe(Ⅱ)、Co(Ⅱ)、Ni(Ⅱ)等金属的配合物已被证实能够在适当的光化学条件下将二氧化碳分子还原为一氧化碳及甲酸[6-10]。尤其进入新世纪以来,由于能源和环境问题日益突出,基于廉价金属Fe(Ⅱ)和Co(Ⅱ)配合物的光催化二氧化碳还原研究得到特别关注并不断取得进展[9, 10]。限于篇幅,本文将回顾以金属钴配合物作为催化剂的均相光催化二氧化碳还原分子体系的发展,总结用于光催化二氧化碳还原研究的钴金属配合物的类型,重点讨论钴配合物光催化二氧化碳还原研究的新进展。
1 钴-氮杂四/五元环类配合物将金属钴配合物作为催化剂进行光催化CO2还原的研究始于上世纪80年代初。1982年,法国科学家Lehn等人报道了以CoCl2为催化剂、Ru(bpy)32+为光敏剂、三异丙基胺(NPr3)为电子牺牲体的可见光催化CO2还原体系[11],体系能够在可见光照射下将溶解于乙腈/水混合溶剂中的CO2分子还原为CO并同时生成了H2,虽然体系的催化效率不高(基于Co2+计算的CO和H2的TON加和值仅为9),但这一研究初步揭示了二价钴离子可在上述光催化体系中将CO2催化还原为CO。在体系中进一步加入联吡啶配体能够增加体系生成气体(CO + H2)的总量,但是降低了CO的选择性。
随后,基于二价钴的氮杂四元环类配合物C1~C4相继被报道能够在光催化体系中将CO2还原[12-15], 见图 2。其中,在以Ru(bpy)32+为光敏剂、抗坏血酸(H2A)为电子牺牲体的水溶液中,配合物C1和C2均能将CO2还原为CO,其在最优条件下的TON(CO)值分别为22和38[12],但是由于体系构筑于水溶液中,光催化析氢反应不可避免,含有C1和C2的体系对CO的选择性分别仅为22%和30%。

|
图 2 配合物C1~C5的结构 Fig.2 The structures of complexes C1-C5 |
基于二价钴的氮杂四元环类配合物中,Co-cyclam配合物(C3)的研究受到较多关注[13-15]。1993年,Yanagida研究组报道了C3能够在以对三联苯(TP)为光敏剂、三乙胺(TEA)为电子牺牲体的紫外光照射体系中将CO2还原为CO,并有少量甲酸根离子(HCOO-)生成[13]。该小组的后续研究发现,用吩嗪替换体系中的对三联苯[14],体系的产物分布发生了改变,甲酸根离子成为CO2还原的主产物,作者推测在该体系中,由激发态吩嗪到催化剂之间的光诱导电子转移过程同时伴随着质子迁移,生成了[CoL(H)]2+中间体(L为配体)——这种金属氢化物有助于催化CO2向甲酸根离子的转化。
2015年,Lau和Robert研究组发现二价钴的氮杂五元环配合物C5(结构见图 2)能高效在以Ir(ppy)3为光敏剂、三乙胺为电子牺牲体的可见光催化体系中将CO2还原为CO,光照22 h后体系的TON值可达到270,CO的选择性高达97%[16]。电化学及光谱研究表明,体系发生了由激发态光敏剂向C5的光诱导电子转移,生成的两电子还原的[CoI(L·-)]物种作为活性中间体进一步结合CO2分子实现了催化转化。
2 钴-卟啉及类似物1998年至2002年间,Neta研究组和Fujita研究组对金属钴卟啉及其类似物(钴酞菁、钴咕啉、钴咔咯)在光照条件下催化CO2还原的行为进行了系统的研究并报道了系列研究成果[17-21]。他们首先发现钴卟啉C6(见图 3)在含有三乙胺的乙腈溶液中光照(λ>320 nm)能够催化还原CO2生成CO和HCOO-,其总TON值(CO + HCOO-)大于300,但随着光照时间的延长,钴卟啉逐渐分解导致体系失活[17]。作者认为,体系中生成的零价钴中间体Co0TPP是催化CO2还原的活性物种,而在上述体系中,Co0TPP中间体由两个CoITPP物种发生歧化反应生成,因此量子产率不高。为了提高Co0TPP中间体的量子产率,他们将对三联苯(TP)引入体系中,光照后激发态TP与三乙胺之间发生还原性猝灭生成的TP·-能进一步将一价钴卟啉(CoITPP)还原为零价(Co0TPP);与不含TP的体系相比,Co0TPP中间体的量子产率及催化还原CO2为CO的效率均明显提升[18]。

|
图 3 配合物C6~C9的结构 Fig.3 The structures of complexes C6-C9 |
随后,Neta研究组相继报道了钴酞菁(C7)[19]、钴咕啉(C8)[20]、钴咔咯(C9)[21]3种钴卟啉类似物光催化还原CO2的实例(结构见图 3)。该系列研究表明,上述3种钴卟啉类似物均能在以TP为光敏剂、TEA为电子牺牲体的有机溶剂(DMF或CH3CN)中将CO2还原为CO及甲酸根离子,同时有不同程度的H2产生。其光催化反应机理类似:TP受光激发后与TEA之间发生还原性猝灭,生成高活性的TP·-并进一步将钴金属配合物还原,还原态的钴金属配合物结合CO2分子进行后续催化转化。对于以钴酞菁C7为催化剂的体系,其光催化还原CO2为CO的TON值为50;对以钴咕啉C8为催化剂的体系,其光催化还原CO2为CO和甲酸根离子的TON值大于100;以钴咔咯C9为催化剂的体系,其光催化还原CO2为CO和甲酸根离子的TON值在50~100之间。上述体系虽然能够实现光催化CO2还原,但是由于使用了TP作为光敏剂,因此需要使用紫外光照射才能完成光催化。
3 钴-多吡啶配合物1999年,Kasuga研究组[22]报道了一例含Ru(Ⅱ)和Co(Ⅲ)的双金属核二元分子光催化剂模型C10和C11(见图 4),其结构中的Ru配合物单元和Co配合物单元分别作为光敏剂和催化剂。作者发现将Co配合物单元键连到Ru配合物单元形成双核配合物后,Ru配合物单元的发光被有效猝灭,而在同样条件下的分子间体系的发光并无明显猝灭现象。以C10或C11为光催化剂、三乙醇胺(TEOA)为电子牺牲体在DMF/水混合溶液中构建的光催化CO2还原体系光照29 h后,产物中有CO、H2和甲酸生成,CO为主要的气相产物,其中C10和C11体系生成CO的TON值分别约为3和5。同样光照条件下,以[Co(bpy)3]3+(C12)为催化剂、[Ru(bpy)3]Cl2为光敏剂组成的分子间体系其光催化还原CO2为CO的TON值约为8.7,但体系的主要气相产物为氢气。

|
图 4 配合物C10~C12的结构 Fig.4 The structures of complexes C10-C12 |
2003年,Hirose等人[23]以[Co(bpy)3]2+为催化剂、[Ru(bpy)3]2+为光敏剂、TEOA为电子牺牲体在DMF中构筑了光催化CO2还原体系,与Kasuga体系不同的是,该体系中[Ru(bpy)3]2+被固定在Nifion树脂上以期增加光敏剂的稳定性。体系光照长达400 h后,生成的CO的转换数仅为8.3,且CO/H2的比率仅为0.45。
2015年,Chan等人[24]发现五配位的二价钴配合物[Co(TPA)Cl]+(C13)(见图 5)能够在以Ir(ppy)3为光敏剂、三乙胺为电子牺牲体的乙腈溶液中实现可见光催化还原CO2。三组分体系光照70 h后,体系生成了CO和H2,其中CO的TON值达到953,选择性达到85%。
最近,Lau和Robert研究组[25]发现,四齿吡啶配体与Co2+螯合形成的配合物C14(图 5)能够在光催化体系中将CO2有效还原为CO。该光催化体系以[Ru(bpy)3]2+为光敏剂、C14为催化剂、BIH为电子牺牲体在乙腈/TEOA混合溶剂中构筑,体系在蓝色LED灯的照射下能够高效将CO2还原为CO,其TON值高达2660,对CO的选择性达到98%。令人欣喜的是,将贵金属光敏剂[Ru(bpy)3]2+替换为有机染料Purpurin后,体系CO的转化数仍可保持在790——这是目前不含贵金属的均相光催化CO2还原体系达到的最高转化数值。

|
图 5 配合物C13、C14、电子牺牲体BIH及光敏剂Purpurin的结构 Fig.5 The chemical structures of C13, C14, BIH and Purpurin |
最近几年,以金属钴配合物为催化剂进行光催化CO2还原的研究得到进一步发展,开发新型钴配合物催化剂、从催化剂结构层面认识CO2还原机理成为了研究的主要趋势。
前期报道的绝大多数钴配合物催化剂均采用tran-构型,这使得在催化循环过程中CO2分子与金属中心的结合一般发生在配合物的轴向配体。2016年,Che研究组[26]首次报道了一系列结构新颖的具有cis-构型的二价钴配合物C15~C18(图 6)——配合物中两个氯原子占据两个相邻的cis配位点。研究表明,该系列配合物均能在电催化、光催化体系中将CO2还原为CO,其中活性最高的C15电催化还原CO2的法拉第效率高达96%;在Ir(ppy)3作为光敏剂、三乙胺作为电子牺牲体组成的光催化体系中,生成CO的TON值和选择性可达到368和95%。实验及理论计算表明,在催化还原CO2的过程中,C15催化剂上cis位的一个氯原子首先离开,之后该cis位被CO2分子占据并结合一个质子后形成[Co(COOH)Cl]加合物,而另一cis位的Cl配体能够与结合在金属中心上的COOH部分形成有效的分子内氢键。与两个氯配体均离去的[Co(COOH)]加合物相比,cis位的氯配体与二氧化碳之间形成的分子内氢键能有效降低反应中间体的活化能。这一结果表明cis构型的钴配合物其相邻的两个cis能够在CO2还原中协同工作,从而降低还原过程的活化能,提升催化剂的催化活性。

|
图 6 配合物C15~C18的结构 Fig.6 The structures of complexes C15-C18 |
最近,中山大学鲁统部教授课题组报道了一个双核金属钴配合物C19[27](图 7),与结构类似的单核钴配合物C20相比,C19能够在以[Ru(phen)3]2+为光敏剂、TEOA为电子牺牲体的光催化条件下高效地将CO2催化转化为CO,其生成CO的TON值和选择性高达16896和98%;而在同样条件下,以单核配合物C20为催化剂的体系其生成CO的TON值和选择性仅为1600和85%。DFT计算表明,双核配合物的高催化活性归因于催化还原CO2的过程中双核配合物中两个金属钴原子的协同作用。这是目前以金属钴配合物为催化剂的可见光催化还原二氧化碳的最高TON报道值。

|
图 7 配合物C19和C20的结构 Fig.7 The structures of complexes C19 and C20 |
综上所述,经过近四十年的发展,科学家们陆续发现钴-氮杂四/五元环配合物、钴-卟啉类及类似物、钴-多吡啶类配合物等能够在一定的光催化条件下催化还原CO2,生成CO或/和甲酸根。这一时期的研究拓展了钴配合物催化剂的种类,但绝大多数体系的光催化CO2还原效率和选择性并不高,且部分体系由于使用有机分子作为光敏剂,需要在紫外光的照射下工作。近年来,基于钴配合物的可见光催化CO2还原研究再次受到关注,从分子层面设计新型配合物催化剂,理解催化剂结构与催化机理之间的内在联系成为当前研究的主要趋势。在此基础上发展出的几例钴配合物催化剂表现出优异的光催化CO2还原活性及选择性。但是纵观已报道的基于钴配合物的光催化二氧化碳还原体系,笔者认为以下几个问题仍需进一步关注:首先,体系的转化数、选择性及稳定性仍需要进一步提高;其次,大多数体系仍使用贵金属作为光敏剂;再次,光催化循环机理,尤其是CO2在配合物中心的转化机理仍有待阐明;最后,已报道的体系对CO2的还原程度不高,其产物均为两电子还原产物CO或甲酸,因此从催化剂分子设计和光催化体系设计的角度提升分子催化剂对CO2的还原程度仍是未来面临的挑战。进一步理解光合作用系统的工作原理以及CO2在催化剂金属中心的转化历程,从分子层面有的放矢地设计合成高活性、高选择性、高稳定性的配合物催化剂,结合新型功能材料构筑高效、稳定、还原程度及仿生程度高的可见光催化二氧化碳还原体系,将成为该领域发展的方向。
| [1] | Berardi S, Drouet S, Francàs L, Gimbert-Suriñach C, Guttentag M, Richmond C, Stoll T, Llobet A. Molecular artificial photosynthesis[J]. Chemical Society Reviews, 2014, 43(22): 7501–7519. DOI:10.1039/C3CS60405E |
| [2] | Tran P D, Wong L H, Barber J, Loo J S C. Recent advances in hybrid photocatalysts for solar fuel production[J]. Energy & Environmental Science, 2012, 5(3): 5902–5918. |
| [3] | Shi J F, Jiang Y J, Jiang Z Y, Wang X Y, Wa ng, X L, Zhang S H, Han P P, Yang C. Enzymatic conversion of carbon dioxide[J]. Chemical Society Reviews, 2015, 44(17): 5981–6000. DOI:10.1039/C5CS00182J |
| [4] | Liu X, Inagaki S, Gong J L. Heterogeneous molecular systems for photocatalytic CO2 reduction with water oxidation[J]. Angewandte Chemie International Edition, 2016, 55(48): 14924–14950. DOI:10.1002/anie.201600395 |
| [5] | Aresta M, Dibenedetto A, Angelini A. Catalysis for the valorization of exhaust carbon:from CO2 to chemicals, mate-rials, and fuels. Technological use of CO2[J]. Chemical Reviews, 2013, 114(3): 1709–1742. |
| [6] | Cokoja M, Bruckmeier C, Rieger B, Herrmann W A, Kühn F E. Transformation of carbon dioxide with homogeneous transition-metal catalysts:a molecular solution to a global challenge?[J]. Angewandte Chemie International Edition, 2011, 50(37): 8510–8537. DOI:10.1002/anie.201102010 |
| [7] | Takeda H, Ishitani O. Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies[J]. Coordination Chemistry Reviews, 2010, 254(3): 346–354. |
| [8] | Morris A J, Meyer G J, Fujita E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels[J]. Accounts of Chemical Research, 2009, 42(12): 1983–1994. DOI:10.1021/ar9001679 |
| [9] | Takeda H, Cometto C, Ishitani O, Robert M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction[J]. ACS Catalysis, 2017, 7(1): 70–88. DOI:10.1021/acscatal.6b02181 |
| [10] | Bonin J, Maurin A, Robert M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances[J]. Coordination Chemistry Reviews, 2017, 334: 184–198. DOI:10.1016/j.ccr.2016.09.005 |
| [11] | Lehn J M, Ziessel R. Photochemical generation of carbon monoxide and hydrogen by reduction of carbon dioxide and water under visible light irradiation[J]. Proceedings of the National Academy of Sciences, 1982, 79(2): 701–704. DOI:10.1073/pnas.79.2.701 |
| [12] | Tinnemans A H A, Koster T P M, Thewissen D H M W, Mackor A. Tetraaza-macrocyclic cobalt (Ⅱ) and nickel (Ⅱ) complexes as electron-transfer agents in the photo (electro) chemical and electrochemical reduction of carbon dioxide[J]. Recueil des Travaux Chimiques des Pays-Bas, 1984, 103(10): 288–295. |
| [13] | Matsuoka S, Yamamoto K, Ogata T, Kusaba M, Nakashima N, Fujita E, Yanagida S. Efficient and selective electron mediation of cobalt complexes with cyclam and related macrocycles in the p-terphenyl-catalyzed photoreduction of carbon dioxide[J]. Journal of the American Chemical Society, 1993, 115(2): 601–609. DOI:10.1021/ja00055a032 |
| [14] | Ogata T, Yamamoto Y, Wada Y, Murakoshi K, Kusaba M, Nakashima N, Ishida A, Takamuku S, Yanagida S. Phenazine-photosensitized reduction of CO2 mediated by a cobalt-cyclam complex through electron and hydrogen transfer[J]. The Journal of Physical Chemistry, 1995, 99(31): 11916–11922. DOI:10.1021/j100031a020 |
| [15] | Ogata T, Yanagida S, Brunschwig B S, Fujita E. Mechanistic and kinetic studies of cobalt macrocycles in a photochemical CO2 reduction system:Evidence of Co-CO2 adducts as intermediates[J]. Journal of the American Chemi-cal Society, 1995, 117(25): 6708–6716. DOI:10.1021/ja00130a009 |
| [16] | Chen L, Guo Z, Wei X G, Gallenkamp C, Bonin J, Anxolabéhère-Mallart E, Lau K C, Lau T C, Robert M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with earth-abundant metal complexes. Selective production of CO vs HCOOH by switching of the metal center[J]. Journal of the American Chemical Society, 2015, 137(34): 10918–10921. DOI:10.1021/jacs.5b06535 |
| [17] | Behar D, Dhanasekaran T, Neta P, Hosten C M, Ejeh D, Hambright P, Fujita E. Cobalt porphyrin catalyzed reduction of CO2. Radiation chemical, photochemical, and electrochemical studies[J]. The Journal of Physical Chemistry A, 1998, 102(17): 2870–2877. DOI:10.1021/jp9807017 |
| [18] | Dhanasekaran T, Grodkowski J, Neta P, Hambright P, Fujita E. P-terphenyl-sensitized photoreduction of CO2 with cobalt and iron porphyrins. Interaction between CO and reduced metalloporphyrins[J]. The Journal of Physical Chemistry A, 1999, 10(38): 7742–7748. |
| [19] | Grodkowski J, Dhanasekaran T, Neta P, Hambright P, Brunschwig B S, Shinozaki K, Fujita E. Reduction of cobalt and iron phthalocyanines and the role of the reduced species in catalyzed photoreduction of CO2[J]. The Journal of Physical Chemistry A, 2000, 104(48): 11332–11339. DOI:10.1021/jp002709y |
| [20] | Grodkowski J, Neta P. Cobalt corrin catalyzed photoreduction of CO2[J]. The Journal of Physical Chemistry A, 2000, 104(9): 1848–1853. DOI:10.1021/jp9939569 |
| [21] | Grodkowski J, Neta P, Fujita E, Mahammed A, Simkho-vich L, Gross Z. Reduction of cobalt and iron corroles and catalyzed reduction of CO2[J]. The Journal of Physical Chemistry A, 2002, 106(18): 4772–4778. DOI:10.1021/jp013668o |
| [22] | Komatsuzaki N, Himeda Y, Hirose T, Sugihara H, Kasuga K. Synthesis and photochemical properties of Ruthenium-Cobalt and Ruthenium-Nickel dinuclear complexes[J]. Bulletin of the Chemical Society of Japan, 1999, 72(4): 725–731. DOI:10.1246/bcsj.72.725 |
| [23] | Hirose T, Maeno Y, Himeda Y. Photocatalytic carbon dioxide photoreduction by Co(bpy)32+ sensitized by Ru (bpy)32+ fixed to cation exchange polymer[J]. Journal of Molecular Catalysis A:Chemical, 2003, 193(1): 27–32. |
| [24] | Chan S L F, Lam T L, Yang C, Yan S C, Cheng N M. A robust and efficient cobalt molecular catalyst for CO2 reduction[J]. Chemical Communications, 2015, 51(37): 7799–7801. DOI:10.1039/C5CC00566C |
| [25] | Guo Z, Cheng S, Cometto C, Anxolabéhère-Mallart E, Ng S M, Ko C C, Liu G J, Chen L J, Robert M, Lau T C. Highly efficient and selective photocatalytic CO2 reduction by iron and cobalt quaterpyridine complexes[J]. Journal of the American Chemical Society, 2016, 138(30): 9413–9416. DOI:10.1021/jacs.6b06002 |
| [26] | Wang F, Cao B, To W P, Tse C W, Li K, Chang X Y, Zang C, Chan S L F, Che C M. The effects of chelating N4 ligand coordination on Co (ii)-catalysed photochemical conversion of CO2 to CO:reaction mechanism and DFT calculations[J]. Catalysis Science & Technology, 2016, 6(20): 7408–7420. |
| [27] | Ouyang T, Huang H H, Wang J W, Zhong D C, Lu T B. A dinuclear cobalt cryptate as a homogeneous photocatalyst for highly selective and efficient visible-light driven CO2 reduction to CO in CH3CN/H2O solution[J]. Angewandte Chemie International Edition, 2016, 129(3): 756–761. |